Formulation of Liver-Specific PLGA-DY-635 Nanoparticles Loaded with the Protein Kinase C Inhibitor Bisindolylmaleimide I

 ,
,  ,
,  , ,
, ,
Abstract
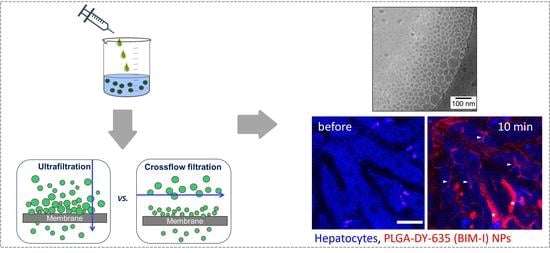
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Nanoparticle Formulation
2.3. Dynamic Light Scattering (DLS)
2.4. Cryo Transmission Electron Microscopy (cryo-TEM)
2.5. Analytical Ultracentrifugation (AUC)
2.6. UV–VIS Spectroscopy
2.7. High-Performance Liquid Chromatography (HPLC)
2.8. Cell Viability Assay
2.9. Animals and Intravital Microscopy
2.10. Phospho-PKC Substrate Western Blot
3. Results and Discussion
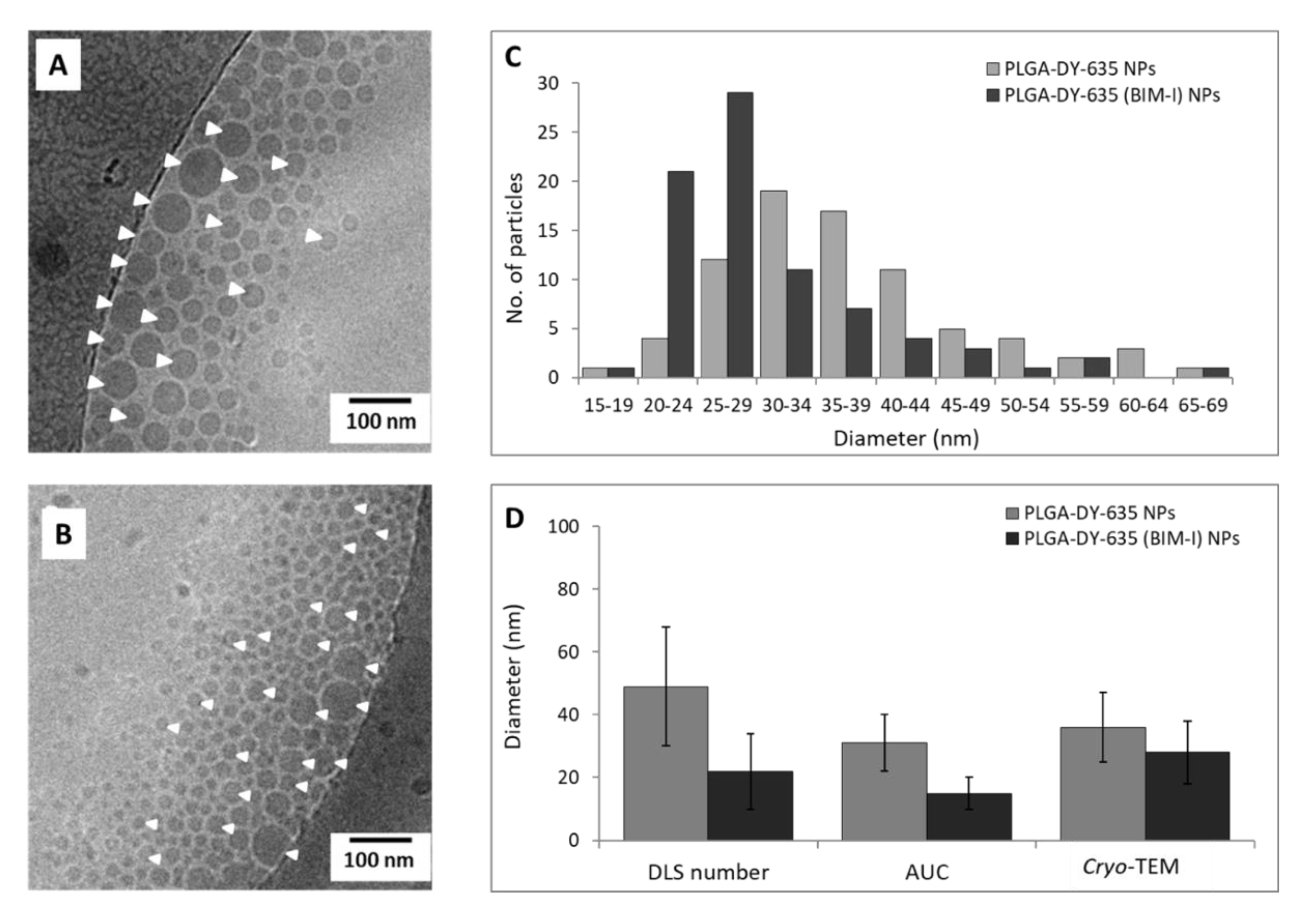
3.1. Nanoparticle Characterization
Size, Population Distribution, and Surface Charge
3.2. Influence of the Purification Method on the Encapsulation Efficiency
3.3. Effect of the Particles on Cell Viability
3.4. Uptake of NPs in Hepatocytes In Vivo
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Toullec, D.; Pianetti, P.; Coste, H.; Bellevergue, P.; Grand-Perret, T.; Ajakane, M.; Baudet, V.; Boissin, P.; Boursier, E.; Loriolle, F. The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. J. Biol. Chem. 1991, 266, 15771–15781. [Google Scholar] [PubMed]
- Hers, I.; Tavaré, J.M.; Denton, R.M. The protein kinase C inhibitors bisindolylmaleimide I (GF 109203x) and IX (Ro 31-8220) are potent inhibitors of glycogen synthase kinase-3 activity. FEBS Lett. 1999, 460, 433–436. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, Y.; Huang, F.L.; Nakabayashi, H.; Huang, K.-P. Tissue distribution and developmental expression of protein kinase C isozymes. J. Biol. Chem. 1988, 263, 9868–9873. [Google Scholar] [PubMed]
- Mochly-Rosen, D.; Kauvar, L.M. Modulating Protein Kinase C. Adv. Pharmacol. 1998, 44, 91–145. [Google Scholar]
- Black, A.R.; Black, J.D. Protein kinase C signaling and cell cycle regulation. Front. Immunol. 2012, 3, 423. [Google Scholar] [CrossRef] [Green Version]
- Schmitz-Peiffer, C.; Biden, T.J. Protein kinase C function in muscle, liver, and β-cells and its therapeutic implications for type 2 diabetes. Diabetes 2008, 57, 1774–1783. [Google Scholar] [CrossRef] [Green Version]
- Di Mari, J.F.; Mifflin, R.C.; Powell, D.W. The role of protein kinase C in gastrointestinal function and disease. Gastroenterology 2005, 128, 2131–2146. [Google Scholar] [CrossRef]
- Ramm, G.A.; Li, L.; Britton, R.S.; O’Neill, R.; Bacon, B.R. Effect of protein kinase C activation and inhibition on rat hepatic stellate cell activation. Dig. Dis. Sci. 2003, 48, 790–796. [Google Scholar] [CrossRef]
- Yang, J.I.; Yoon, J.-H.; Bang, Y.-J.; Lee, S.-H.; Lee, S.-M.; Byun, H.J.; Myung, S.-J.; Kim, W.; Lee, H.-S. Synergistic antifibrotic efficacy of statin and protein kinase C inhibitor in hepatic fibrosis. Am. J. Physiol. Liver Physiol. 2010, 298, G126–G132. [Google Scholar] [CrossRef]
- Crocenzi, F.A.; Sánchez Pozzi, E.J.; Ruiz, M.L.; Zucchetti, A.E.; Roma, M.G.; Mottino, A.D.; Vore, M. Ca2+-dependent protein kinase C isoforms are critical to estradiol 17β-D-glucuronide-induced cholestasis in the rat. Hepatology 2008, 48, 1885–1895. [Google Scholar] [CrossRef] [Green Version]
- Corasanti, J.G.; Smith, N.D.; Gordon, E.R.; Boyer, J.L. Protein kinase C agonists inhibit bile secretion independently of effects on the microcirculation in the isolated perfused rat liver. Hepatology 1989, 10, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Kubitz, R. Protein kinase C–dependent distribution of the multidrug resistance protein 2 from the canalicular to the basolateral membrane in human HepG2 cells. Hepatology 2001, 34, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Anwer, M.S. Role of protein kinase C isoforms in bile formation and cholestasis. Hepatology 2014, 60, 1090–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobson, P.B.; Kuchera, S.L.; Metz, A.; Schächtele, C.; Imre, K.; Schrier, D.J. Anti-inflammatory properties of Gö 6850: A selective inhibitor of protein kinase C. J. Pharmacol. Exp. Ther. 1995, 275, 995–1002. [Google Scholar] [PubMed]
- Komander, D.; Kular, G.S.; Schüttelkopf, A.W.; Deak, M.; Prakash, K.R.C.; Bain, J.; Elliott, M.; Garrido-Franco, M.; Kozikowski, A.P.; Alessi, D.R.; et al. Interactions of LY333531 and other bisindolyl maleimide inhibitors with PDK1. Structure 2004, 12, 215–226. [Google Scholar] [CrossRef]
- Scortegagna, M.; Ruller, C.; Feng, Y.; Lazova, R.; Kluger, H.; Li, J.L.; De, S.K.; Rickert, R.; Pellecchia, M.; Bosenberg, M.; et al. Genetic inactivation or pharmacological inhibition of Pdk1 delays development and inhibits metastasis of Braf V600E::Pten -/- melanoma. Oncogene 2014, 33, 4330–4339. [Google Scholar] [CrossRef] [Green Version]
- Gekeler, V.; Boer, R.; Uberall, F.; Ise, W.; Schubert, C.; Utz, I.; Hofmann, J.; Sanders, K.H.; Schächtele, C.; Klemm, K.; et al. Effects of the selective bisindolylmaleimide protein kinase C inhibitor GP 109203X on P-glycoprotein-mediated multidrug resistance. Br. J. Cancer 1996, 74, 897–905. [Google Scholar] [CrossRef] [Green Version]
- Kosgodage, U.S.; Trindade, R.P.; Thompson, P.R.; Inal, J.M.; Lange, S. Chloramidine/bisindolylmaleimide-I-mediated inhibition of exosome and microvesicle release and enhanced efficacy of cancer chemotherapy. Int. J. Mol. Sci. 2017, 18, 1007. [Google Scholar] [CrossRef]
- Stone, R.M.; Manley, P.W.; Larson, R.A.; Capdeville, R. Midostaurin: Its odyssey from discovery to approval for treating acute myeloid leukemia and advanced systemic mastocytosis. Blood Adv. 2018, 2, 444–453. [Google Scholar] [CrossRef] [Green Version]
- Pinter, M.; Trauner, M.; Peck-Radosavljevic, M.; Sieghart, W. Cancer and liver cirrhosis: Implications on prognosis and management. ESMO Open 2016, 1, e42. [Google Scholar] [CrossRef]
- Tapper, E.B.; Parikh, N.D. Mortality due to cirrhosis and liver cancer in the United States, 1999–2016: Observational study. BMJ 2018, 362, k2817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jepsen, P. Comorbidity in cirrhosis. World J. Gastroenterol. 2014, 20, 7223–7230. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.; Hammerling, B.C.; Wimmer, A.-B.; Wu, K.; Ficker, E.; Kuryshev, Y.A.; Scherer, D.; Kiehn, J.; Katus, H.A.; Schoels, W.; et al. Direct block of hERG potassium channels by the protein kinase C inhibitor bisindolylmaleimide I (GF109203X). Cardiovasc. Res. 2004, 64, 467–476. [Google Scholar] [CrossRef]
- Harmati, G.; Papp, F.; Szentandrássy, N.; Bárándi, L.; Ruzsnavszky, F.; Horváth, B.; Bányász, T.; Magyar, J.; Panyi, G.; Krasznai, Z.; et al. Effects of the PKC inhibitors chelerythrine and bisindolylmaleimide I (GF 109203X) on delayed rectifier K+ currents. Naunyn. Schmiedebergs. Arch. Pharmacol. 2011, 383, 141–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ertl, P.; Rohde, B.; Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef]
- Shkodra-Pula, B.; Grune, C.; Traeger, A.; Vollrath, A.; Schubert, S.; Fischer, D.; Schubert, U.S. Effect of surfactant on the size and stability of PLGA nanoparticles encapsulating a protein kinase C inhibitor. Int. J. Pharm. 2019, 566, 756–764. [Google Scholar] [CrossRef]
- Hoshyar, N.; Gray, S.; Han, H.; Bao, G. The effect of nanoparticle size on in vivo pharmacokinetics and cellular interaction. Nanomedicine 2016, 11, 673–692. [Google Scholar] [CrossRef] [Green Version]
- Press, A.T.; Traeger, A.; Pietsch, C.; Mosig, A.; Wagner, M.; Clemens, M.G.; Jbeily, N.; Koch, N.; Gottschaldt, M.; Bézière, N.; et al. Cell type-specific delivery of short interfering RNAs by dye-functionalised theranostic nanoparticles. Nat. Commun. 2014, 5, 5565. [Google Scholar] [CrossRef] [Green Version]
- Thakkar, N.; Lockhart, A.C.; Lee, W. Role of irganic anion-transporting polypeptides (OATPs) in cancer therapy. AAPS J. 2015, 17, 535–545. [Google Scholar] [CrossRef] [Green Version]
- Stetefeld, J.; McKenna, S.A.; Patel, T.R. Dynamic light scattering: A practical guide and applications in biomedical sciences. Biophys. Rev. 2016, 8, 409–427. [Google Scholar] [CrossRef]
- Mie, G. Beiträge zur Optik trüber Medien, speziell kolloidaler Metallösungen. Ann. Phys. 1908, 330, 377–445. [Google Scholar] [CrossRef]
- Schuck, P.; Rossmanith, P. Determination of the sedimentation coefficient distribution by least-squares boundary modeling. Biopolymers 2000, 54, 328–341. [Google Scholar] [CrossRef]
- Cinar, G.; Englert, C.; Lehmann, M.; Nischang, I. In situ quantitative assessment of multifunctional nanoscale drug delivery systems in human serum. Anal. Chem. 2020, 92, 7932–7939. [Google Scholar] [CrossRef] [PubMed]
- Spek, S.; Haeuser, M.; Schaefer, M.M.; Langer, K. Characterisation of PEGylated PLGA nanoparticles comparing the nanoparticle bulk to the particle surface using UV-VIS spectroscopy, SEC, 1H-NMR spectroscopy, and X-ray photoelectron spectroscopy. Appl. Surf. Sci. 2015, 347, 378–385. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasband, W.S.; ImageJ, U.S. National Institutes of Health, Bethesda, Maryland, USA. Available online: https://imagej.nih.gov/ij/docs/faqs.html (accessed on 16 November 2020).
- Agrawal, A.; Rellegadla, S.; Jain, S. Biomedical applications of PLGA particles. In Materials for Biomedical Engineering; Holban, A.-M., Grumezescu, A.M., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 87–129. [Google Scholar]
- Dhakar, R.C.; Maurya, S.D.; Sagar, B.P.S.; Bhagat, S.; Kumar, P.S.; Jain, C.P. Variables influencing the drug entrapment efficiency of microspheres: A pharmaceutical review. Der Pharm. Lett. 2010, 2, 102–116. [Google Scholar]
- Huang, W.; Zhang, C. Tuning the size of poly(lactic-co-glycolic acid) (PLGA) nanoparticles fabricated by nanoprecipitation. Biotechnol. J. 2018, 13, 1700203. [Google Scholar] [CrossRef]
- Schubert, S.; Delaney, J.T., Jr.; Schubert, U.S. Nanoprecipitation and nanoformulation of polymers: From history to powerful possibilities beyond poly(lactic acid). Soft Matter 2011, 7, 1581–1588. [Google Scholar] [CrossRef]
- Beck-Broichsitter, M.; Nicolas, J.; Couvreur, P. Solvent selection causes remarkable shifts of the “Ouzo region” for poly(lactide-co-glycolide) nanoparticles prepared by nanoprecipitation. Nanoscale 2015, 7, 9215–9221. [Google Scholar] [CrossRef]
- Shkodra-Pula, B.; Vollrath, A.; Schubert, U.S.; Schubert, S. Polymer-based nanoparticles for biomedical applications. In Colloids for Nanobiotechnology: Synthesis, Characterization and Potential; Wolfgang, J.P., Feliu, N., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 233–252. ISBN 9780081028285. [Google Scholar]
- Italia, J.L.; Sharp, A.; Carter, K.C.; Warn, P.; Kumar, M.N.V.R. Peroral amphotericin B polymer nanoparticles lead to comparable or superior in vivo antifungal activity to that of intravenous Ambisome® or FungizoneTM. PLoS ONE 2011, 6, e25744. [Google Scholar] [CrossRef] [Green Version]
- Karnieli, O. Bioreactors and downstream processing for stem cell manufacturing. In Stem Cell Manufacturing; Elsevier: Amsterdam, The Netherlands, 2016; pp. 141–160. ISBN 9780444632654. [Google Scholar]
- Bhattacharjee, S. DLS and zeta potential—What they are and what they are not? J. Control. Release 2016, 235, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Wyatt Technology. What are Some Considerations and Strategies When Working with Fluorescing Samples? Available online: https://www.wyatt.com/support/ask-the-expert-what-are-some-considerations-and-strategies-when-working-with-fluorescing-samples.html (accessed on 17 January 2020).
- Finsy, R.; de Jaeger, N.; Sneyers, R.; Geladé, E. Particle sizing by photon correlation spectroscopy. Part III: Mono and bimodal distributions and data analysis. Part. Part. Syst. Charact. 1992, 9, 125–137. [Google Scholar] [CrossRef]
- Nickel, C.; Angelstorf, J.; Bienert, R.; Burkart, C.; Gabsch, S.; Giebner, S.; Haase, A.; Hellack, B.; Hollert, H.; Hund-Rinke, K.; et al. Dynamic light-scattering measurement comparability of nanomaterial suspensions. J. Nanopart. Res. 2014, 16, 2260. [Google Scholar] [CrossRef]
- Franks, K.; Kestens, V.; Braun, A.; Roebben, G.; Linsinger, T.P.J. Non-equivalence of different evaluation algorithms to derive mean particle size from dynamic light scattering data. J. Nanopart. Res. 2019, 21, 195. [Google Scholar] [CrossRef] [Green Version]
- Sharma, N.; Madan, P.; Lin, S. Effect of process and formulation variables on the preparation of parenteral paclitaxel-loaded biodegradable polymeric nanoparticles: A co-surfactant study. Asian J. Pharm. Sci. 2016, 11, 404–416. [Google Scholar] [CrossRef] [Green Version]
- Sharma, D.; Maheshwari, D.; Philip, G.; Rana, R.; Bhatia, S.; Singh, M.; Gabrani, R.; Sharma, S.K.; Ali, J.; Sharma, R.K.; et al. Formulation and optimization of polymeric nanoparticles for intranasal delivery of lorazepam using Box-Behnken design: In vitro and in vivo evaluation. Biomed Res. Int. 2014, 2014, 156010. [Google Scholar] [CrossRef] [Green Version]
- Reisch, A.; Runser, A.; Arntz, Y.; Mély, Y.; Klymchenko, A.S. Charge-controlled nanoprecipitation as a modular approach to ultrasmall polymer nanocarriers: Making bright and stable nanoparticles. ACS Nano 2015, 9, 5104–5116. [Google Scholar] [CrossRef]
- Gumustas, M.; Sengel-Turk, C.T.; Gumustas, A.; Ozkan, S.A.; Uslu, B. Effect of polymer-based nanoparticles on the assay of antimicrobial drug delivery systems. In Multifunctional Systems for Combined Delivery, Biosensing and Diagnostics; Elsevier: Amsterdam, The Netherlands, 2017; pp. 67–108. ISBN 9780323527255. [Google Scholar]
- Ruiz-García, A.; Melián-Martel, N.; Nuez, I. Short review on predicting fouling in RO desalination. Membranes 2017, 7, 62. [Google Scholar] [CrossRef] [Green Version]
- Shakesheff, K.M.; Evora, C.; Soriano, I.; Langer, R. The adsorption of poly(vinyl alcohol) to biodegradable microparticles studied by X-ray photoelectron spectroscopy (XPS). J. Colloid Interface Sci. 1997, 185, 538–547. [Google Scholar] [CrossRef]
- Arora, M. Cell culture media: A Review. Mater. Methods 2013, 3, 1–29. [Google Scholar] [CrossRef]
- Schöttler, S.; Klein, K.; Landfester, K.; Mailänder, V. Protein source and choice of anticoagulant decisively affect nanoparticle protein corona and cellular uptake. Nanoscale 2016, 8, 5526–5536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behzadi, S.; Serpooshan, V.; Tao, W.; Hamaly, M.A.; Alkawareek, M.Y.; Dreaden, E.C.; Brown, D.; Alkilany, A.M.; Farokhzad, O.C.; Mahmoudi, M. Cellular uptake of nanoparticles: Journey inside the cell. Chem. Soc. Rev. 2017, 46, 4218–4244. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, R.; Cicalese, A.; Fumagalli, M.; Dobreva, M.; Verrecchia, A.; Pelicci, P.G.; Di Fagagna, F.D.A. DNA damage response activation in mouse embryonic fibroblasts undergoing replicative senescence and following spontaneous immortalization. Cell Cycle 2008, 7, 3601–3606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fröhlich, E. Comparison of conventional and advanced in vitro models in the toxicity testing of nanoparticles. Artif. Cells Nanomed. Biotechnol. 2018, 46, 1091–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prats-Mateu, B.; Ertl, P.; Toca-Herrera, J.L. Influence of HepG2 cell shape on nanoparticle uptake. Microsc. Res. Tech. 2014, 77, 560–565. [Google Scholar] [CrossRef] [PubMed]
- Menon, J.U.; Kona, S.; Wadajkar, A.S.; Desai, F.; Vadla, A.; Nguyen, K.T. Effects of surfactants on the properties of PLGA nanoparticles. J. Biomed. Mater. Res. Part A 2012, 100A, 1998–2005. [Google Scholar] [CrossRef]
- Liu, T.; Mi, L.; Xiong, J.; Orchard, P.; Yu, Q.; Yu, L.; Zhao, X.Y.; Meng, Z.X.; Parker, S.C.J.; Lin, J.D.; et al. BAF60a deficiency uncouples chromatin accessibility and cold sensitivity from white fat browning. Nat. Commun. 2020, 11, 1–15. [Google Scholar]
- Cell Signaling Technology Phospho-MAPK/CDK Substrates (PXS*P or S*PXR/K) (34B2) Rabbit mAb (Sepharose® Bead Conjugate). Available online: https://www.cellsignal.de/datasheet.jsp?productId=5501&images=1 (accessed on 3 November 2020).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
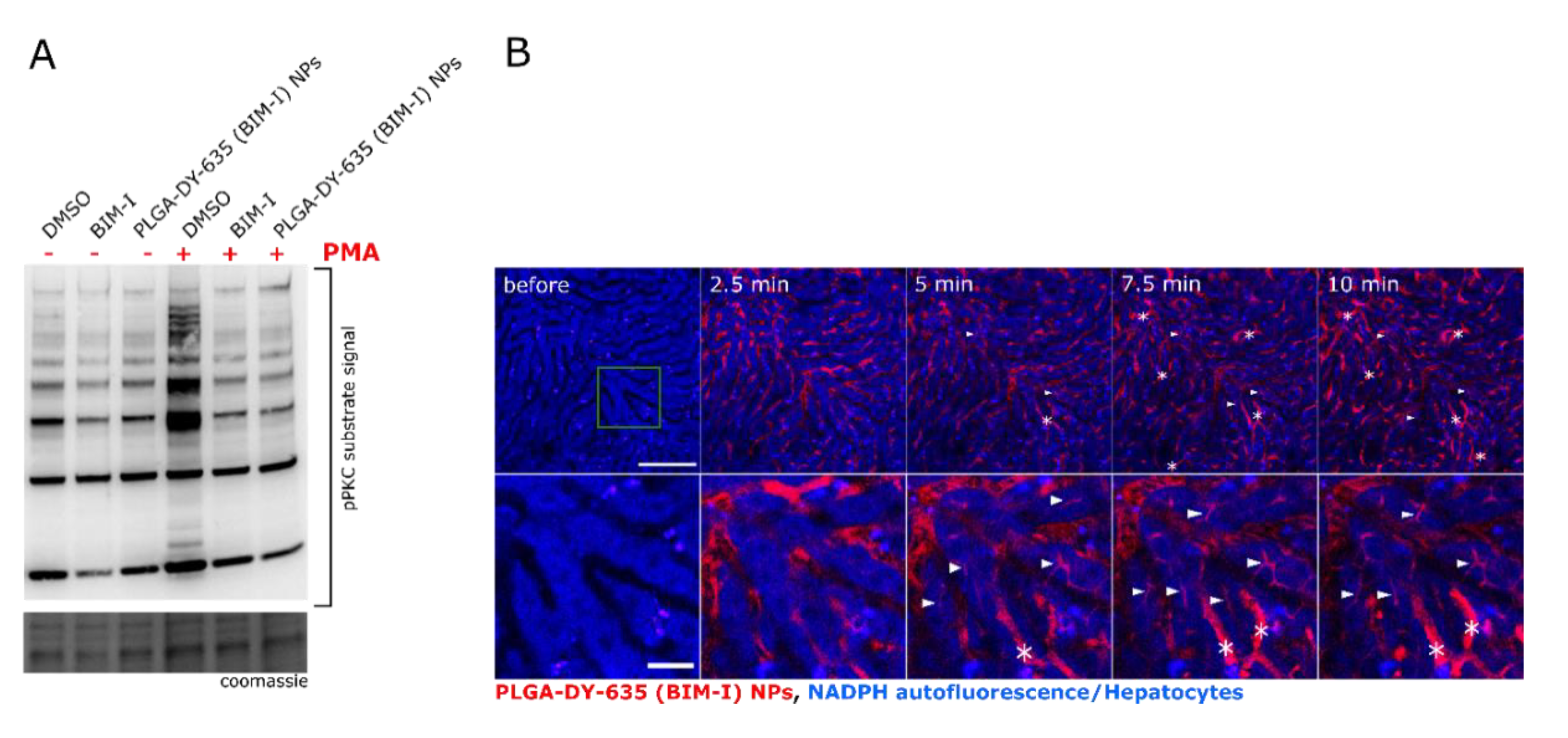{kind=link}
| NP Formulation | Purification Method | dH (nm) a ± SD | dH (nm) b ± SD | dH (nm) c ± SD | PDI ± SD | ζ (mV) ± SD |
|---|---|---|---|---|---|---|
| PLGA-DY-635_U | Ultrafiltration | 120 ± 24 | 133 ± 72 | 66 ± 19 | 0.15 ± 0.07 | −20 ± 8 |
| PLGA-DY-635_C | Crossflow filtration | 65 ± 17 | 50 ± 26 | 49 ± 3 | 0.15 ± 0.03 | −20 ± 0 |
| PLGA-DY-635 (BIM-I)_U | Ultrafiltration | 92 ± 3 | 73 ± 2 | 28 ± 8 | 0.22 ± 0.05 | −7 ± 10 |
| PLGA-DY-635 (BIM-I)_C | Crossflow filtration | 63 ± 7 | 37 ± 3 | 22 ± 2 | 0.30 ± 0.08 | −9 ± 2 |
| NP Formulation | BIM-I a (µg mL−1) ± SD | BIM-I b (µg mL−1) ± SD | EE (%) ± SD | LC (%) ± SD | NP + PVA (mg mL−1) ± SD | PVA (%, w/v) ± SD |
|---|---|---|---|---|---|---|
| PLGA-DY-635_U | / | / | / | / | 18 ± 1 | 1.1 ± 0.1 |
| PLGA-DY-635_C | / | / | / | / | 23 | 0.9 |
| PLGA-DY-635 (BIM-I)_U | 192 ± 15 | 194 ± 42 | 32 ± 18 | 1.5 ± 1 | 25 ± 1 | 1.5 ± 0.2 |
| PLGA-DY-635 (BIM-I)_C | 419 | 419 | 60 | 3 | 24 | 1.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shkodra, B.; Press, A.T.; Vollrath, A.; Nischang, I.; Schubert, S.; Hoeppener, S.; Haas, D.; Enzensperger, C.; Lehmann, M.; Babic, P.; et al. Formulation of Liver-Specific PLGA-DY-635 Nanoparticles Loaded with the Protein Kinase C Inhibitor Bisindolylmaleimide I. Pharmaceutics 2020, 12, 1110. https://doi.org/10.3390/pharmaceutics12111110
Shkodra B, Press AT, Vollrath A, Nischang I, Schubert S, Hoeppener S, Haas D, Enzensperger C, Lehmann M, Babic P, et al. Formulation of Liver-Specific PLGA-DY-635 Nanoparticles Loaded with the Protein Kinase C Inhibitor Bisindolylmaleimide I. Pharmaceutics. 2020; 12(11):1110. https://doi.org/10.3390/pharmaceutics12111110
Chicago/Turabian StyleShkodra, Blerina, Adrian T. Press, Antje Vollrath, Ivo Nischang, Stephanie Schubert, Stephanie Hoeppener, Dorothee Haas, Christoph Enzensperger, Marc Lehmann, Petra Babic, and et al. 2020. "Formulation of Liver-Specific PLGA-DY-635 Nanoparticles Loaded with the Protein Kinase C Inhibitor Bisindolylmaleimide I" Pharmaceutics 12, no. 11: 1110. https://doi.org/10.3390/pharmaceutics12111110