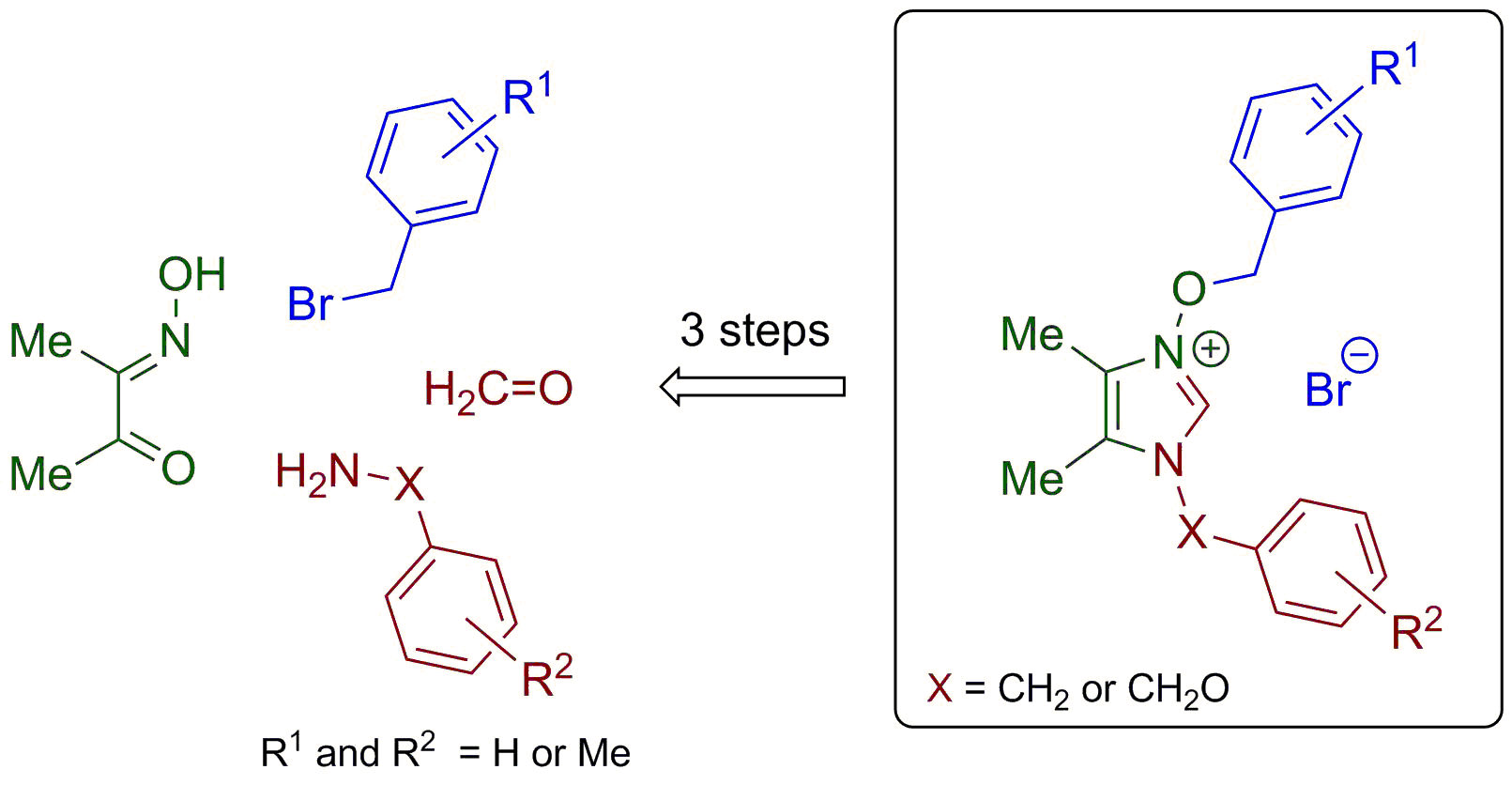
2.1.1. Synthesis of Imidazole N-Oxides 7 and 8
Method A: To a solution of diacetyl monooxime (2a, 505 mg, 5.0 mmol) or benzyl monooxime (2b, 1.12 g, 5.0 mmol) in glacial acetic acid (15 mL) was added appropriate formaldimine 1 (5.0 mmol), and the resulting mixture was stirred at room temperature overnight. Then, excess concentrated hydrochloric acid was added (0.2 mL), the solvents were removed under reduced pressure, the resulting was dissolved in methanol (100 mL), excess solid NaHCO3 (ca. 5.0 g) was added, and the stirring was continued for ca. 30 min until the evolution of CO2 ceased. After the crude organic salt was fully neutralized, the solvent was removed in vacuo and the residue was triturated with dichloromethane (30 mL). The precipitate was filtered off and the solvent was evaporated to give imidazole N-oxide 3, which was either further purified by column chromatography or recrystallization from a diisopropyl ether/dichloromethane mixture. As per the literature, known imidazole N-oxides 3a–b,g–i crude products were washed with a portion of diethyl ether (ca. 30 mL) and used as received. Analytically pure samples were obtained by crystallization from a diisopropyl ether/dichloromethane mixture (slow evaporation at room temperature).
Method B: A mixture of equimolar amounts of α-hydroxyiminoketone of type 2 (5.0 mmol) and corresponding formaldimine 1 (5.0 mmol) in EtOH (10 mL) was refluxed for 4 h. The solvent was removed, and the resulting oily material was triturated with several portions of diethyl ether (4 × 15 mL). The resulting crude imidazole N-oxides 3 were purified by recrystallization from diisopropyl ether/dichloromethane mixture (slow evaporation at room temperature).
1-Benzyl-4,5-dimethyl-1
H-imidazole 3-oxide (
3a):
Method B: 880 mg (87%). Colorless solid, melting point (m.p.) 200–201 °C (199–201 °C [
19]).
1H-NMR (CDCl
3, 600 MHz): δ 2.07, 2.20 (2 s, 3 H each, 2 Me), 5.00 (s, 2 H), 7.08–7.11, 7.31–7.38 (2 m, 2 H, 3 H, Bn), 7.88 (s
br, 1 H, C(2)H) ppm.
1-Benzyl-4,5-diphenyl-1
H-imidazole 3-oxide (
3b):
Method B: 1.32 g (81%). Colorless solid, m.p. 176–177 °C (176–178 °C [
19]).
1H-NMR (CDCl
3, 600 MHz): δ 4.93 (s, 2 H), 7.03–7.05, 7.18–7.42, 7.55–7.58 (3 m, 2 H, 11 H, 2 H, 3 Ph), 7.98 (s, 1 H, C(2)H) ppm.
1-Benzyloxy-4,5-dimethyl-1H-imidazole 3-oxide (3c): Method A: 719 mg (66%); Method B: 0%. Crude product was purified by column chromatography (SiO2, AcOEt/MeOH 1:1, Rf = 0.5) to give 7d as colorless solid, m.p. 103–105 °C. 1H-NMR (CDCl3, 600 MHz): δ 1.94, 2.10 (2 s, 3 H each, 2 Me), 5.03 (s, 2 H, Bn), 7.27–7.29, 7.35–7.42 (2 m, 2 H, 3 H, Bn), 7.73 (sbr, 1 H, C(2)H) ppm. 13C-NMR (CDCl3, 151 MHz): δ 7.0, 7.2 (2 q, 2 Me), 82.7 (t, Bn), 119.3 (s, Im), 120.6 (d, C(2)), 123.7 (s, Im), 129.0, 129.9, 130.1 (3 d, Bn), 132.4 (s, Bn) ppm. IR (neat): ν 3070, 1675, 1457, 1390, 1172, 1079, 941, 908 cm−1. Electrospray ionization (ESI)–MS (m/z): 241.2 (42, [M + Na]+), 219.3 (100, [M + H]+). C12H14N2O2·0.8 H2O: calculated, C 61.95, H 6.76, N 12.04; found, C 61.90, H 6.84, N 12.10.
1-(2-Methylbenzyloxy)-4,5-dimethyl-1H-imidazole 3-oxide (3d): Method A: 709 mg (61%). Colorless solid, m.p. 89–91 °C. 1H-NMR (CDCl3, 600 MHz): δ 1.91, 2.07, 2.36 (3 s, 3 H each, 3 Me), 5.07 (s, 2 H, CH2), 7.05–7.07, 7.11–7.14, 7.19–7.21, 7.25–7.29 (4 m, 1 H each), 7.70 (s, 1 H, C(2)H) ppm. 13C-NMR (CDCl3, 151 MHz): δ 6.9, 7.1, 18.8 (3 q, 3 Me), 80.9 (t, CH2), 119.3 (s, Im), 120.5 (d, C(2)), 123.6 (s, Im), 126.4, 130.4 (2 d, 2 CH), 130.6 (s, i-C), 130.8, 131.2 (2 d, 2 CH), 138.0 (s, i-C) ppm. IR (neat): ν 1444, 1351, 1169, 1079, 922, 744 cm−1. ESI-MS (m/z): 255.1 (88, [M + Na]+), 233.1 (100, [M + H]+). C13H16N2O2·2H2O (268.3): calcdulated, C 58.19, H 7.51, N 10.44; found, C 58.34, H 6.78, N 10.64.
1-(4-Methylbenzyloxy)-4,5-dimethyl-1H-imidazole 3-oxide (3e): Method A: 789 mg (68%). Colorless solid, m.p. 101–102 °C. 1H-NMR (CDCl3, 600 MHz): δ 1.93, 2.08, 2.32 (3 s, 3 H each, 3 Me), 4.96 (s, 2 H, CH2), 7.14 (mc, 4 H), 7.65 (s, 1 H, C(2)H) ppm. 13C-NMR (CDCl3, 151 MHz): δ 6.9, 7.2, 21.2 (3 q, 3 Me), 82.5 (t, CH2), 119.2 (s, Im), 120.6 (d, C(2)), 123.5 (s, Im), 129.4 (s, i-C), 129.6, 129.9 (2 d, 4 CH), 140.2 (s, i-C) ppm. IR (neat): ν 1601, 1448, 1318, 1299, 1172, 1170, 922 cm−1. ESI-MS (m/z): 255.0 (46, [M + Na]+), 233.1 (100, [M + H]+). C13H16N2O2·H2O: (250.29): calculated, C 62.38, H 7.25, N 11.19; found, C 62.06, H 7.07, N 11.60.
1-(3,5-Dimethylbenzyloxy)-4,5-dimethyl-1H-imidazole 3-oxide (3f): Method A: 1.17 g (95%). Colorless solid, m.p. 96–98 °C. 1H-NMR (CDCl3, 600 MHz): δ 1.98, 2.10 (2 s, 3 H each, 2 Me), 2.27 (s, 6 H, 2 Me), 4.94 (s, 2 H, CH2), 6.88 (sbr, 2 H), 7.01 (sbr, 1 H), 7.69 (s, 1 H, C(2)H) ppm. 13C-NMR (CDCl3, 151 MHz): δ 6.9, 7.2 (2 q, 2 Me), 21.1 (q, 2 Me), 82.9 (t, CH2), 119.2 (s, Im), 120.6 (d, C(2)), 123.6 (s, Im), 127.4 (d, 2 CH), 131.6 (d, CH), 132.3 (s, i-C), 138.7 (s, 2 i-C) ppm. IR (neat): ν 1608, 1394, 1172, 1081, 938 cm−1. ESI-MS (m/z): 285.1 (100, [M + K]+), 269.1 (54, [M + Na]+), 247.1 (87, [M + H]+). C14H18N2O2·1.3 H2O: calculated, C 62.20, H 7.71, N 10.36; found, C 62.14, H 7.53, N 10.30.
1-Adamantyl-4,5-dimethyl-1
H-imidazole 3-oxide (
3g):
Method A: 677 mg (55%). Colorless solid, m.p. 179–180 °C (decomposed) (m.p. 180–182 °C (decomposed) [
20]).
1H-NMR (CDCl
3, 600 MHz): δ 1.72, 1.77 (2 d
br,
J ≈ 12.5 Hz, 6 H, Ad), 2.13 (m
c, 6 H, Ad), 2.17 (s, 3 H, Me), 2.24 (m
c, 3 H, Ad), 2.36 (s, 3 H, Me), 7.89 (s, 1 H, C(2)H) ppm.
1-Adamantyl-4,5-diphenyl-1
H-imidazole 3-oxide (
3h):
Method A: 814 mg (44%). Colorless solid, m.p. 234–239 °C (decomposed) (m.p. 238–241 °C (decomposed) [
20]).
1H-NMR (CDCl
3, 600 MHz): δ 1.54, 1.65 (2 d
br,
J ≈ 12.2 Hz, 6 H, Ad), 2.05 (m
c, 6 H, Ad), 2.11 (m
c, 3 H, Ad), 7.14–7.21, 7.33–7.50 (2 m, 3 H, 7 H, 2 Ph), 8.21 (s, 1 H, C(2)H) ppm.
1-Adamantyloxy-4,5-dimethyl-1
H-imidazole 3-oxide (
3i):
Method A: 968 mg (74%). Pale yellow solid, m.p. 103–106 °C (m.p. 104–106 °C [
17]).
1H-NMR (CDCl
3, 600 MHz): δ 1.59, 1.70 (2 d
br,
J ≈ 12.3 Hz, 6 H, Ad), 1.86 (m
c, 6 H, Ad), 2.16, 2.20 (2 s, 3 H each, 2 Me), 2.27 (m
c, 3 H, Ad), 7.85 (s, 1 H, C(2)H) ppm.
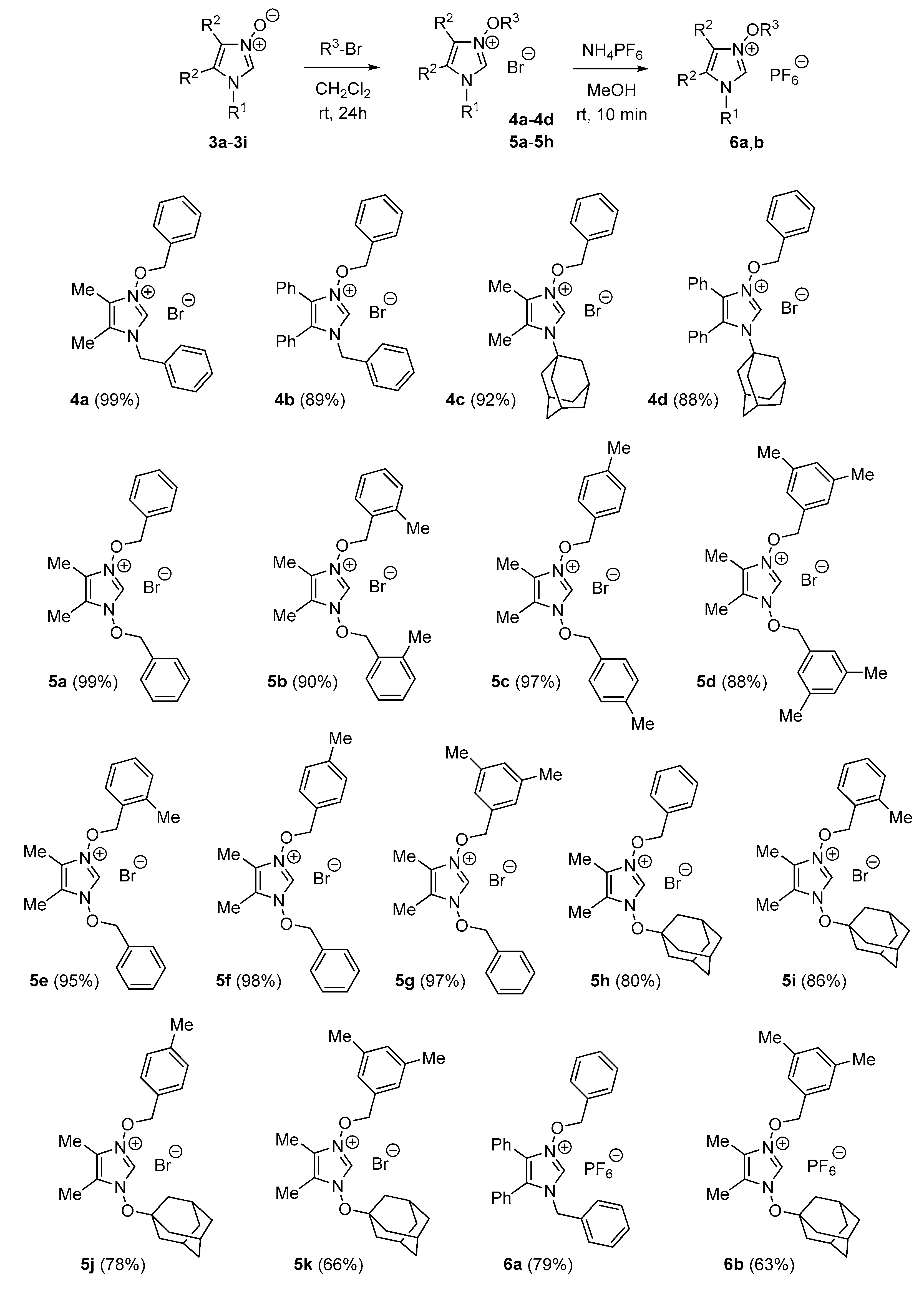
2.1.2. General Procedure for the Synthesis of Imidazolium Bromides 4 and 5
To a solution of corresponding imidazole N-oxide 3 (1.0 mmol) in dry dichloromethane (2.0 mL) was added excess alkyl bromide (5.0 mL), and the resulting mixture was stirred at rt until the starting N-oxide was fully consumed (thin-layer chromatography (TLC) monitoring: SiO2, EtOAc/MeOH 6:1; typically 24–48 h). After the solvent was removed under reduced pressure, the resulting crude product was triturated with several portions of Et2O (4 × 10 mL) in order to remove excess of unconsumed alkylating agent. The product was dried under high vacuum to give the corresponding imidazolium bromides, whose identity was confirmed by NMR spectroscopy. Analytically pure samples of products 4 and 5 were obtained by crystallization from diisopropyl ether/dichloromethane mixture (slow evaporation at room temperature).
1-Benzyl-3-benzyloxy-4,5-dimethylimidazolium bromide (4a): 369 mg (99%). Colorless solid, m.p. 148–150 °C. 1H-NMR (CDCl3, 600 MHz): δ 1.92, 2.07 (2 s, 3 H each, 2 Me), 5.52, 5.57 (2 s, 2 H each, 2 Bn), 7.28–7.42, 7.49–7.52 (2 m, 8 H, 2 H, 2 Bn), 11.00 (s, 1 H, C(2)H) ppm. 13C-NMR (CDCl3, 151 MHz): δ 7.1, 8.9 (2 q, 2 Me), 51.3 (t, NBn), 84.0 (t, OBn), 124.1, 124.8 (2 s, Im), 128.0, 129.9, 129.0, 129.2, 130.3, 130.6 (6 d, 2 Bn), 131.5 (s, Bn), 132.5 (dbr, C(2)), 132.9 (s, Bn) ppm. IR (neat): ν 2924, 1453, 1340, 1139, 909 cm−1. C19H21N2OBr (373.3): calculated, C 61.13, H 5.67, N 7.50; found, C 61.08, H 5.73, N 7.69.
1-Benzyl-3-benzyloxy-4,5-diphenylimidazolium bromide (4b): 442 mg (89%). Colorless solid, m.p. 167–169 °C. 1H-NMR (CDCl3, 600 MHz): δ 5.42 (s, 2 H, Bn), 5.58 (s, 2 H, Bn), 7.11–7.13, 7.18–7.21, 7.25–7.33, 7.39–7.45, 7.51–7.54 (5 m, 2 H, 4 H, 10 H, 3 H, 1 H, 2 Ph, 2 Bn), 11.17 (s, 1 H, C(2)H) ppm. 13C-NMR (CDCl3, 151 MHz): δ 51.3 (t, NBn), 84.3 (t, OBn), 122.9, 124.2 (2 s, Im), 128.61*, 128.63, 128.73, 128.91, 128.94, 130.5, 130.8, 130.9 (8 d, 20 CH, 2 Ph, 2 Bn), 128.72, 129.3, 129.4, 133.1 (4 s, 2 Ph, 2 Bn), 133.7 (dbr, C(2)) ppm; *higher intensity. IR (neat): ν 2861, 1547, 1456, 1385, 1340, 951, 913 cm−1. C29H25N2OBr (497.4): calculated, C 70.02, H 5.07, N 5.63; found, C 69.12, H 5.15, N 5.76.
1-Adamantyl-3-benzyloxy-4,5-dimethylimidazolium bromide (4c): 384 mg (92%). Colorless solid, m.p. 196–197 °C. 1H-NMR (CDCl3, 600 MHz): δ 1.70–1.77 (m, 6 H, Ad), 1.96 (s, 3 H, Me), 2.28 (mc, 3 H, Ad), 2.30–2.33 (m, 6 H, Ad), 2.38 (s, 3 H, Me), 5.77 (s, 2 H, Bn), 7.32–7.37, 7.56–7.59 (2 m, 3 H, 2 H, Bn), 10.44 (s, 1 H, C(2)H) ppm. 13C-NMR (CDCl3, 151 MHz): δ 7.0, 12.4 (2 q, 2 Me), 29.5, 35.2, 41.6 (d, t, t, Ad), 64.0 (s, Ad), 84.0 (t, Bn), 123.1, 126.2 (2 s, Im), 128.7, 129.9, 130.8 (3 d, Bn), 131.8 (dbr, C(2)), 132.2 (s, Bn) ppm. IR (neat): ν 2911, 2853, 1457, 1303, 1224, 1178, 913 cm−1. C22H29N2OBr∙CHCl3∙H2O (554.77): calculated, C 49.79, H 5.81, N 5.05; found, C 50.09, H 5.79, N 5.40.
1-Adamantyl-3-benzyloxy-4,5-diphenylimidazolium bromide (4d): 475 mg (88%). Colorless solid, m.p. 180–182 °C. 1H-NMR (CDCl3, 600 MHz): δ 1.54–1.62 (m, 6 H, Ad), 2.14 (mc, 3 H, Ad), 2.27 (mc, 6 H, Ad), 5.58 (s, 2 H, Bn), 7.10–7.12, 7.19–7.34, 7.37–7.39, 7.44–7.46 (4 m, 2 H, 10 H, 2 H, 1 H, 2 Ph, Bn), 10.79 (s, 1 H, C(2)H) ppm. 13C-NMR (CDCl3, 151 MHz): δ 29.7, 35.0, 42.5 (d, t, t, Ad), 66.5 (s, Ad), 84.0 (t, Bn), 123.1, 127.3 (2 s, Im), 128.4, 128.5, 128.6, 129.68, 129.71, 129.9, 130.6, 131.0, 132.4 (9 d, 2 Ph, Bn), 128.3, 130.4, 131.5 (3 s, 2 Ph, Bn), 132.9 (dbr, C(2)) ppm. IR (neat): ν 2911, 1444, 1157, 911 cm−1. C32H33N2OBr·1.5 CHCl3 (720.58): calculated, C 55.84, H 4.83, N 3.89; found, C 55.70, H 5.24, N 4.45.
1,3-Dibenzyloxy-4,5-dimethylimidazolium bromide (5a): 384 mg (99%). Colorless solid, m.p. 110–111 °C. 1H-NMR (CDCl3, 600 MHz): δ 1.77 (s, 6 H, 2 Me), 5.77 (s, 4 H, 2 Bn), 7.35–7.43, 7.52–7.55 (2 m, 6 H, 4 H, 2 Bn), 11.80 (s, 1 H, C(2)H) ppm. 13C-NMR (CDCl3, 151 MHz): δ 7.0 (q, 2 Me), 84.1 (t, 2 Bn), 122.3 (s, C(4), C(5)), 128.9 (d, 4 CH, 2 Bn), 129.8 (dbr, C(2)), 130.3, 130.8 (2 d, 6 CH, 2 Bn), 131.8 (s, 2 i-C, 2 Bn) ppm. IR (neat): ν 2816, 1623, 1455, 1388, 1215, 1075, 947, 904 cm−1. Crude sample of 5a was transformed into analytically pure imidazole-2-thione 7c (see below).
1,3-Di-(2-methylbenzyloxy)-4,5-dimethylimidazolium bromide (5b): 374 mg (90%). Colorless solid, m.p. 132–133 °C. 1H-NMR (CDCl3, 600 MHz): δ 1.74 (s, 6 H, 2 Me), 2.45 (s, 6 H, 2 Me), 5.75 (s, 4 H, 2 CH2), 7.09–7.11, 7.18–7.20, 7.25–7.28, 7.40–7.43 (4 m, 2 H each), 11.64 (s, 1 H, C(2)H) ppm. 13C-NMR (CDCl3, 151 MHz): δ 6.8 (q, 2 Me), 19.1 (q, 2 Me), 82.6 (t, 2 CH2), 122.3 (s, C(4), C(5)), 126.3 (d, 2 CH), 129.7 (dbr, C(2)), 130.1 (s, 2 i-C), 130.6, 130.7, 132.0 (3 d, 6 CH), 138.7 (s, 2 i-C) ppm. IR (neat): ν 2825, 2691, 1629, 1461, 1440, 1392, 1215, 1081, 922, 871, 749 cm−1. C21H25N2O2Br (417.3): calculated, C 60.44, H 6.04, N 6.71; found, C 60.29, H 5.95, N 7.43.
1,3-Di-(4-methylbenzyloxy)-4,5-dimethylimidazolium bromide (5c): 404 mg (97%). Colorless solid, m.p. 111–113 °C. 1H-NMR (CDCl3, 600 MHz): δ 1.75 (s, 6 H, 2 Me), 2.31 (s, 6 H, 2 Me), 5.64 (s, 4 H, 2 CH2), 7.09–7.12, 7.34–7.36 (2 m, 4 H each), 11.73 (s, 1 H, C(2)H) ppm. 13C-NMR (CDCl3, 151 MHz): δ 7.1 (q, 2 Me), 21.3 (q, 2 Me), 83.9 (t, 2 CH2), 122.2 (s, C(4), C(5)), 128.7 (s, 2 i-C), 129.4 (dbr, C(2)), 129.5, 130.7 (2 d, 8 CH), 140.4 (s, 2 i-C) ppm. IR (neat): ν 2924, 2900, 1625, 1527, 1440, 1381, 1279, 1208, 919, 870, 807 cm−1. C21H25N2O2Br·H2O (435.3): calculated, C 57.94, H 6.25, N 6.43; found, C 57.01, H 6.23, N 6.86.
1,3-Di-(3,5-dimethylbenzyloxy)-4,5-dimethylimidazolium bromide (5d): 391 mg (88%). Colorless solid, m.p. 141–143 °C. 1H-NMR (CDCl3, 600 MHz): δ 1.89 (s, 6 H, 2 Me), 2.24 (s, 12 H, 4 Me), 5.55 (s, 4 H, 2 CH2), 6.99 (sbr, 2 H), 7.06 (sbr, 4 H), 11.62 (s, 1 H, C(2)H) ppm. 13C-NMR (CDCl3, 151 MHz): δ 7.1 (q, 2 Me), 21.0 (q, 4 Me), 84.5 (t, 2 CH2), 122.2 (s, C(4), C(5)), 128.1 (d, 4 CH), 129.6 (dbr, C(2)), 131.4 (s, 2 i-C), 131.8 (d, 2 CH), 138.5 (s, 4 i-C) ppm. IR (neat): ν 2917, 1610, 1459, 1384, 1079, 934, 896 cm−1. C23H29N2O2Br·H2O (463.4): calculated, C 59.61, H 6.74, N 6.05; found, C 59.54, H 7.00, N 6.42.
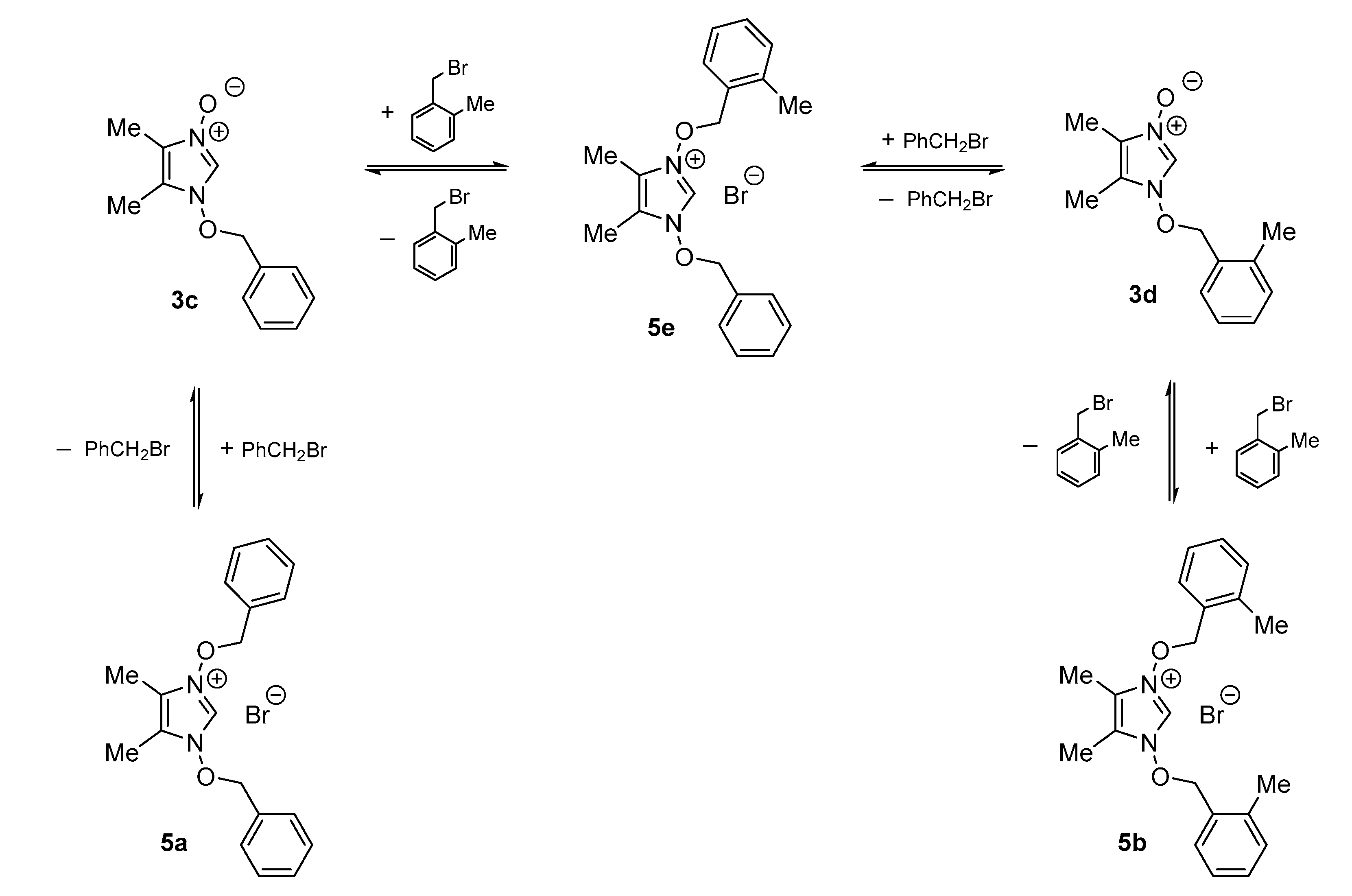
1-Benzyloxy-3-(2-methylbenzyloxy)-4,5-dimethylimidazolium bromide (5e) (in a ca. 20:1:1 mixture with 5a and 5b: yield 383 mg (95%)). Pale yellow oil. 1H-NMR (CDCl3, 600 MHz): δ 1.73, 1.80, 2.46 (3 s, 3 H each, 3 Me), 5.72, 5.73 (2 s, 2 H each, 2 CH2), 7.07–7.10, 7.18–7.21, 7.26–7.30, 7.33–7.40, 7.52–7.55 (5 m, 1 H, 1 H, 1 H, 4 H, 2 H), 11.59 (s, 1 H, C(2)H) ppm. 13C-NMR (CDCl3, 151 MHz): δ 6.8, 7.0, 19.0 (3 q, 3 Me), 82.6, 84.0 (2 t, 2 CH2), 122.3, 122.4 (2 s, Im), 126.2, 128.8 (2 d, 3 CH), 129.3 (dbr, C(2)), 130. (s, i-C), 130.2, 130.57, 130.60, 130.63 (4 d, 5 CH), 131.6 (s, i-C), 131.9 (d, CH), 138.6 (s, i-C) ppm. IR (neat): ν 2924, 1456, 1387, 1215, 1079, 911, 870, 749 cm−1.
1-Benzyloxy-3-(4-methylbenzyloxy)-4,5-dimethylimidazolium bromide (5f): 380 mg (98%). Colorless solid, m.p. 124–126 °C. 1H-NMR (CDCl3, 600 MHz): δ 1.73, 1.74 (2 s, 3 H each, 2 Me), 2.30 (s, 3 H, Me), 5.62, 5.68 (2 s, 2 H each, 2 CH2), 7.09–7.11, 7.25–7.37, 7.45–7.47 (3 m, 2 H, 5 H, 2 H), 11.68 (s, 1 H, C(2)H) ppm. 13C-NMR (CDCl3, 151 MHz): δ 7.04, 7.06 (2 q, 2 Me), 21.3 (q, Me), 83.92, 83.95 (2 t, 2 CH2), 122.2, 122.3 (2 s, Im), 128.6 (s), 128.8 (d, 2 CH), 129.9 (dbr, C(2)), 129.5, 130.2, 130.66, 130.67 (4 d, 7 CH), 131.6, 140.4 (2 s) ppm. IR (neat): ν 2917, 1449, 1375, 1215, 1077, 876 cm−1. C20H23N2O2Br (403.3): calculated, C 59.56, H 5.75, N 6.95; found, C 59.63, H 5.76, N 7.15.
1-Benzyloxy-3-(3,5-dimethylbenzyloxy)-4,5-dimethylimidazolium bromide (5g) (in a 10:1 mixture with 5d: yield 404 mg (97%)). Colorless oil. 1H-NMR (CDCl3, 600 MHz): δ 1.79, 1.85 (2 s, 3 H each, 2 Me), 2.27 (s, 6 H, 2 Me), 5.59, 5.69 (2 s, 2 H each, 2 CH2), 7.01 (sbr, 1 H), 7.09 (sbr, 2 H), 7.30–7.39, 7.46–7.48 (2 m, 3 H, 2 H), 11.64 (s, 1 H, C(2)H) ppm. 13C-NMR (CDCl3, 151 MHz): δ 7.0, 7.1 (2 q, 2 Me), 21.0 (q, 2 Me), 84.2, 84.5 (2 t, 2 CH2), 122.3, 122.4 (2 s, Im), 128.2, 128.9 (2 d, 4 CH), 129.4 (dbr, C(2)), 130.2, 130.6 (2 d, 3 CH), 131.5, 131.7 (2 s, 2 i-C), 131.8 (d, CH), 138.5 (s, 2 i-C) ppm. IR (neat): ν 2923, 1472, 1375, 911, 898 cm−1.
1-Adamantyloxy-3-benzyloxy-4,5-dimethylimidazolium bromide (5h): 346 mg (80%). Colorless solid, m.p. 131–132 °C. 1H-NMR (CDCl3, 600 MHz): δ 1.61-1.69 (m, 6 H, Ad), 1.93 (mc, 6 H, Ad), 2.02, 2.16 (2 s, 3 H each, 2 Me), 2.32 (mc, 3 H, Ad), 5.84 (s, 2 H, Bn), 7.36–7.40, 7.60–7.63 (2 m, 3 H, 2 H, Bn), 11.42 (s, 1 H, C(2)H) ppm. 13C-NMR (CDCl3, 151 MHz): δ 7.4, 8.2 (2 q, 2 Me), 31.2, 35.3, 40.6 (d, t, t, Ad), 84.1 (t, Bn), 91.4 (s, Ad), 122.7, 123.4 (2 s, Im), 128.9, 130.2, 131.0 (3 d, Bn), 131.3 (dbr, C(2)), 131.9 (s, Bn) ppm. IR (neat): ν 2902, 2851, 1638, 1456, 1358, 1217, 1049, 889, cm−1. C22H29N2O2Br∙0.5 H2O (442.4): calculated, C 59.73, H 6.83, N 6.33; found, C 59.54, H 7.00, N 6.42.
1-Adamantyloxy-3-(2-methylbenzyloxy)-4,5-dimethylimidazolium bromide (5i): 374 mg (86%). Colorless solid, m.p. 152–154 °C. 1H-NMR (CDCl3, 600 MHz): δ 1.62–1.68 (m, 6 H, Ad), 1.93 (sbr, 3 H, Me), 1.95–1.98 (m, 6 H, Ad), 2.17 (sbr, 3 H, Me), 2.33 (mc, 3 H, Ad), 2.51 (s, 3 H, Me), 5.88 (s, 2 H, Bn), 7.13–7.15, 7.22–7.24, 7.29–7.33, 7.49–7.51 (4 m, 1 H each), 11.47 (s, 1 H, C(2)H) ppm. 13C-NMR (CDCl3, 151 MHz): δ 7.2, 8.2, 19.2 (3 q, 3 Me), 31.2, 35.3, 40.6 (d, t, t, Ad), 82.8 (t, CH2), 91.5 (s, Ad), 122.9, 123.4 (2 s, Im),126.3 (d, CH), 130.4 (s, i-C), 130.6, 130.7 (2 d, 2 CH), 131.3 (dbr, C(2)), 132.5 (d, CH), 138.7 (s, i-C) ppm. IR (neat): ν 2906, 2849, 1738, 1358, 1216, 1048, 889, 743, cm−1. C23H31N2O2Br·CHCl3 (566.8): calculated, C 50.86, H 5.69, N 4.94; found, C 50.16, H 5.82, N 5.26.
1-Adamantyloxy-3-(4-methylbenzyloxy)-4,5-dimethylimidazolium bromide (5j): 342 mg (78%). Colorless solid, m.p. 130–132 °C. 1H-NMR (CDCl3, 600 MHz): δ 1.59–1.67 (m, 6 H, Ad), 1.92 (mc, 6 H, Ad), 2.01, 2.16 (2 s, 3 H each, 2 Me), 2.33 (mc, 3 H, Ad), 2.33 (s, 3 H, Me), 5.77 (s, 2 H, CH2), 7.15–7.17, 7.46–7.48 (2 m, 2 H each), 11.38 (s, 1 H, C(2)H) ppm. 13C-NMR (CDCl3, 151 MHz): δ 7.4, 8.2, 21.4 (3 q, 3 Me), 31.2, 35.3, 40.6 (d, t, t, Ad), 84.1 (t, CH2), 91.4 (s, Ad), 122.8, 123.3 (2 s, Im), 128.9 (s, i-C), 129.6, 131.0 (2 d, 4 CH), 131.3 (dbr, C(2)), 140.4 (s, i-C) ppm. IR (neat): ν 2911, 2853, 1738, 1378, 1354, 1216, 1043, 879, 813, cm−1. C23H31N2O2Br (447.4): calcd. C 61.74, H 6.98, N 6.26; found: C 61.54, H 7.26, N 6.21.
1-Adamantyloxy-3-(3,5-dimethylbenzyloxy)-4,5-dimethylimidazolium bromide (5k): 306 mg (66%). Pale yellow solid, m.p. 150–152 °C. 1H-NMR (CDCl3, 600 MHz): δ 1.60–1.66 (m, 6 H, Ad), 1.92–1.94 (m, 6 H, Ad), 2.01, 2.16 (2 s, 3 H each, 2 Me), 2.28 (s, 6 H, 2 Me), 2.31 (mc, 3 H, Ad), 5.71 (s, 2 H, CH2), 7.01 (sbr, 1 H), 7.15 (sbr, 2 H), 11.43 (s, 1 H, C(2)H) ppm. 13C-NMR (CDCl3, 151 MHz): δ 7.4, 8.2 (2 q, 2 Me), 21.1 (q, 2 Me), 31.2, 35.3, 40.7 (d, t, t, Ad), 84.6 (t, CH2), 91.4 (s, Ad), 122.8, 123.3 (2 s, Im), 128.5 (d, 2 CH), 131.4 (dbr, C(2)), 131.76 (s, i-C), 131.84 (d, CH), 138.5 (s, 2 i-C) ppm. IR (neat): ν 2910, 2848, 1738, 1359, 1209, 1050, 890, 844 cm−1. C24H33N2O2Br (461.43): calculated, C 62.47, H 7.21, N 6.07; found, C 62.10, H 7.50, N 5.80.
2.1.4. General Procedure for the Synthesis of Imidazole-2-Thiones 7
To a solution of 4,5-dimethylimidazolium bromide of type 4 or 5 (0.50 mmol) in dry pyridine (2.0 mL) was added Et3N (100 μL, 0.75 mmol), followed by a slight excess of elemental sulfur (19.2 mg, 0.60 mmol) at room temperature, and the resulting homogeneous solution was stirred magnetically for 24 h. After removal of solvents in vacuo, the resulting crude products were purified by recrystallization from MeOH to give N-benzyloxy-imidazole-2-thione 7.
1-Benzyl-3-benzyloxy-4,5-dimethylimidazole-2-thione (7a): 122 mg (75%). Colorless crystals, m.p. 116–117 °C (MeOH). 1H-NMR (600 MHz, CDCl3): δ 1.79, 1.87 (2 s, 3 H each, 2 Me), 5.35 (s, 2 H, NCH2), 5.47 (s, 2 H, OCH2), 7.24–7.28, 7.31–7.33, 7.36–7.40, 7.50–7.52 (4 m, 3 H, 2 H, 3 H, 2 H, 2 Bn) ppm. 13C--NMR (CDCl3, 151 MHz): δ 7.4, 9.2 (2 q, 2 Me), 47.8 (t, NCH2), 78.0 (t, OCH2), 117.7, 120.0 (2 s, Im), 127.0, 127.6, 128.6, 128.7, 129.3, 130.4 (6 d, 2 Bn), 133.9, 136.3 (2 s, 2 Bn), 157.4 (s, C=S) ppm. IR (neat): ν 2924, 1403, 1340, 997 cm−1. ESI-MS (m/z): 347.3 (33, [M + Na]+), 325.4 (100, [M + H]+), 293.4 (31). C19H20N2OS (324.1): calculated, C 70.34, H 6.21, N 8.63, S 9.88; found, C 70.24, H 6.28, N 8.77, S 9.79.
1-Adamantyl-3-benzyloxy-4,5-dimethylimidazole-2-thione (7b): 95 mg (52%). Colorless crystals, m.p. 99–101 °C (MeOH). 1H-NMR (CDCl3, 600 MHz): δ 1.67-1.69 (m, 3 H, Ad), 1.82 (s, 3 H, Me), 1.83 (mc, 3 H, Ad), 2.19 (s, 3 H, Me), 2.21 (mc, 3 H, Ad), 2.83–2.85 (m, 6 H, Ad), 5.40 (s, 2 H, Bn), 7.33–7.37, 7.46–7.50 (2 m, 3 H, 2 H, Bn) ppm. 13C-NMR (CDCl3, 151 MHz): δ 7.8, 15.2 (2 q, 2 Me), 30.4, 36.0, 40.6 (d, t, t, Ad), 65.7 (s, Ad), 77.4 (t, Bn), 118.0, 121.0 (2 s, Im), 128.4, 129.1, 130.2 (3 d, Bn), 134.2 (s, Bn), 156.3 (s, C=S) ppm. IR (neat): ν 2913, 2851, 1454, 1360, 1273, 1250, 956 cm−1. ESI-MS (m/z): 369.3 (100, [M + H]+). C22H28N2OS (368.2): calculated, C 71.70, H 7.66, N 7.60, S 8.70; found, C 71.68, H 7.84, N 7.76, S 8.54.
1,3-Dibenzyloxy-4,5-dimethylimidazole-2-thione (7c): 124 mg (73%). Colorless crystals, m.p. 87–88 °C (MeOH). 1H-NMR (600 MHz, CDCl3): δ 1.68 (s, 6 H, 2 Me), 5.45 (s, 4 H, 2 CH2), 7.36–7.40, 7.47–7.59 (2 m, 6 H, 4 H, 2 Bn) ppm. 13C-NMR (CDCl3, 151 MHz): δ 7.3 (q, 2 Me), 78.3 (t, 2 Bn), 116.8 (s, C(4), C(5)), 128.6, 129.4, 130.5 (3 d, 2 Bn), 133.8 (s, 2 Bn), 152.8 (s, C=S) ppm. IR (neat): ν 2917, 1456, 1403, 1384, 1081, 956, 917 cm−1. ESI-MS (m/z): 363.3 (100, [M + Na]+), 341.3 (46, [M + H]+). C19H20N2O2S (340.1): calculated, C 67.03, H 5.92, N 8.23, S 9.42; found, C 67.16, H 5.99, N 8.35, S 9.52.
1-Adamantyloxy-3-benzyloxy-4,5-dimethylimidazole-2-thione (7d): 91 mg (48%). Colorless crystals, m.p. 108–109 °C (MeOH). 1H-NMR (600 MHz, CDCl3): δ 1.65 (mc, 6 H, Ad), 1.76, 2.05 (2 s, 3 H each, 2 Me), 2.15, 2.25 (2 mc, 6 H, 3 H Ad), 5.42 (sbr, 2 H, Bn), 7.35–7.39, 7.47–7.49 (2 m, 3 H, 2 H, Bn) ppm. 13C-NMR (CDCl3, 151 MHz): δ 7.6, 9.4 (2 q, 2 Me), 31.5, 35.9, 42.0 (d, t, t, Ad), 77.9 (t, Bn), 89.0 (s, Ad), 117.2, 118.4 (2 s, Im), 128.5, 129.3, 130.5 (3 d, Bn), 134.0 (s, Bn), 157.0 (s, C=S) ppm. IR (neat): ν 2909, 2850, 1385, 1353, 1045, 945, 906 cm−1. ESI-MS (m/z): 385.2 (100, [M + H]+), 353.2 (93). C22H28N2O2S (384.2): calculated, C 68.72, H 7.34, N 7.29, S 8.34; found, C 68.65, H 7.39, N 7.23, S 8.20.

 ,
, 






{kind=link}
{kind=link}
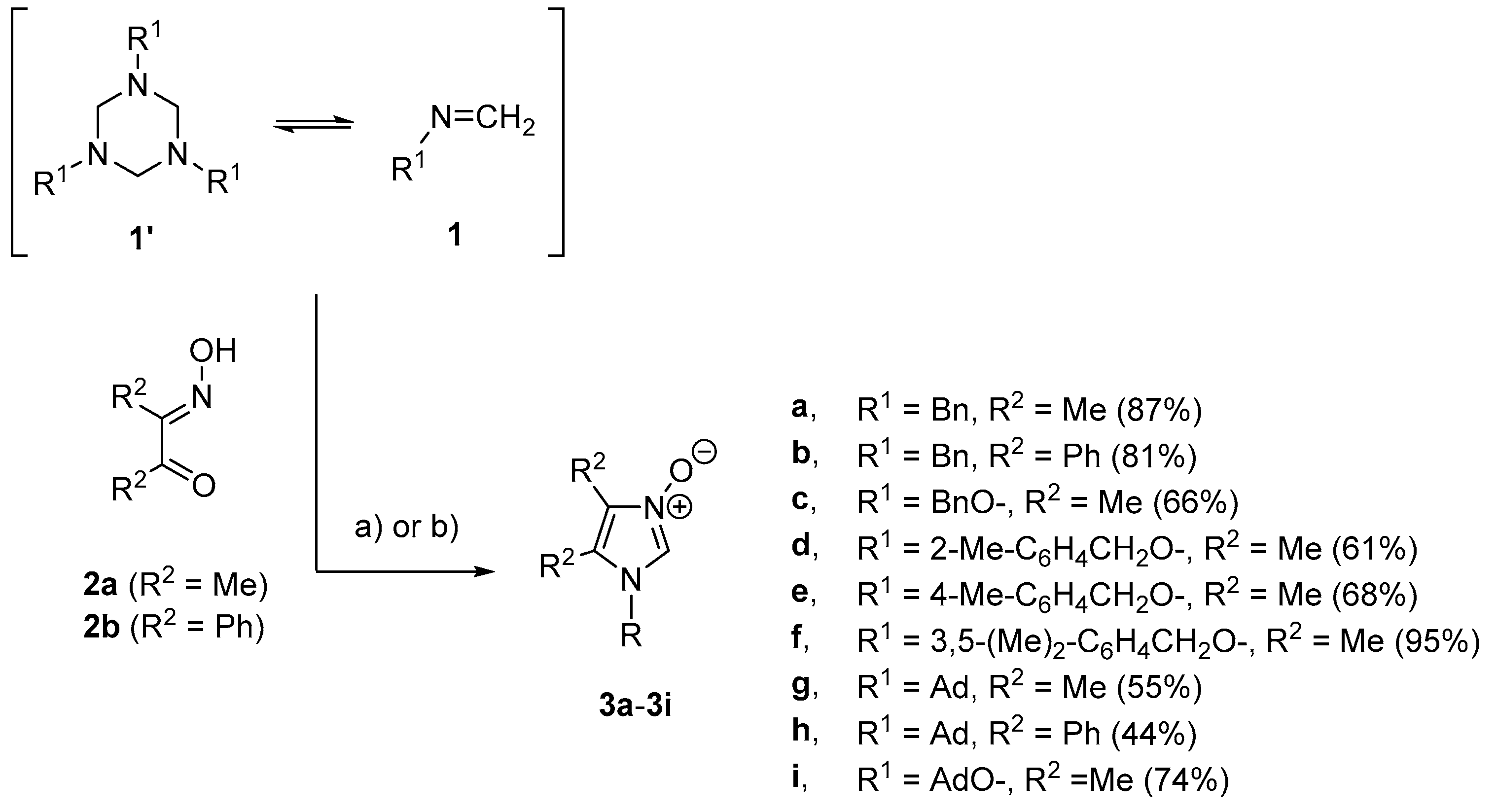{kind=link}
{kind=link}
{kind=link}
{kind=link}
