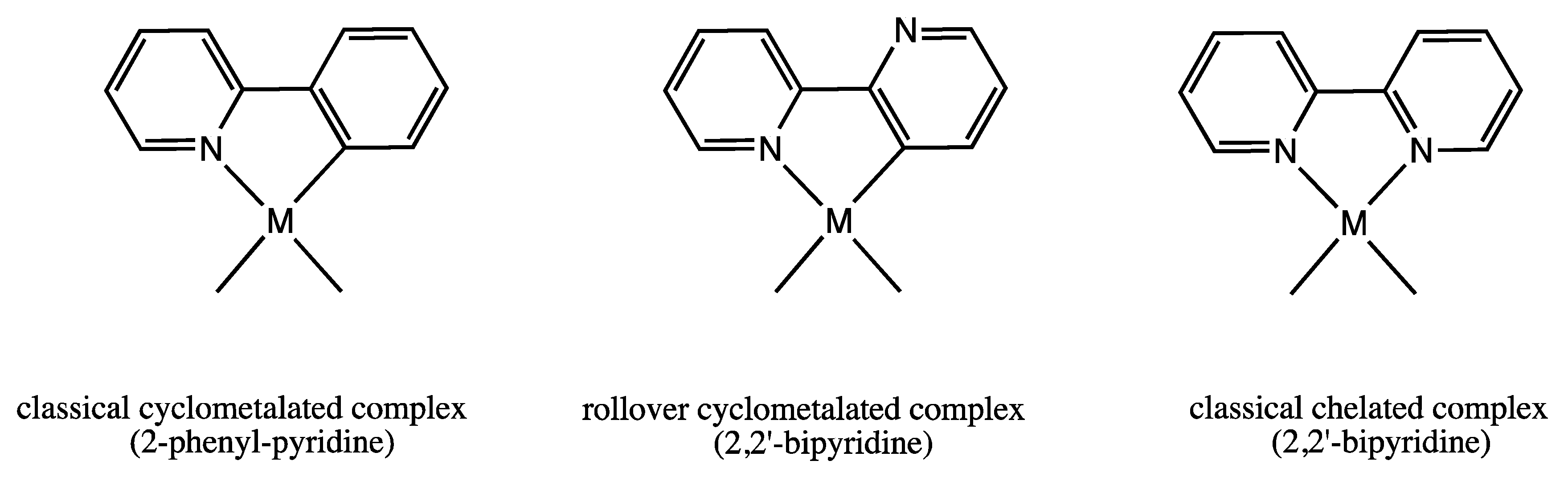
Pt(II) Derivatives with Rollover-Coordinated 6-substituted 2,2′-bipyridines: Ligands with Multiple Personalities

 , , ,
, , ,
Abstract
:Featured Applications
Abstract
1. Introduction

2. Materials and Methods
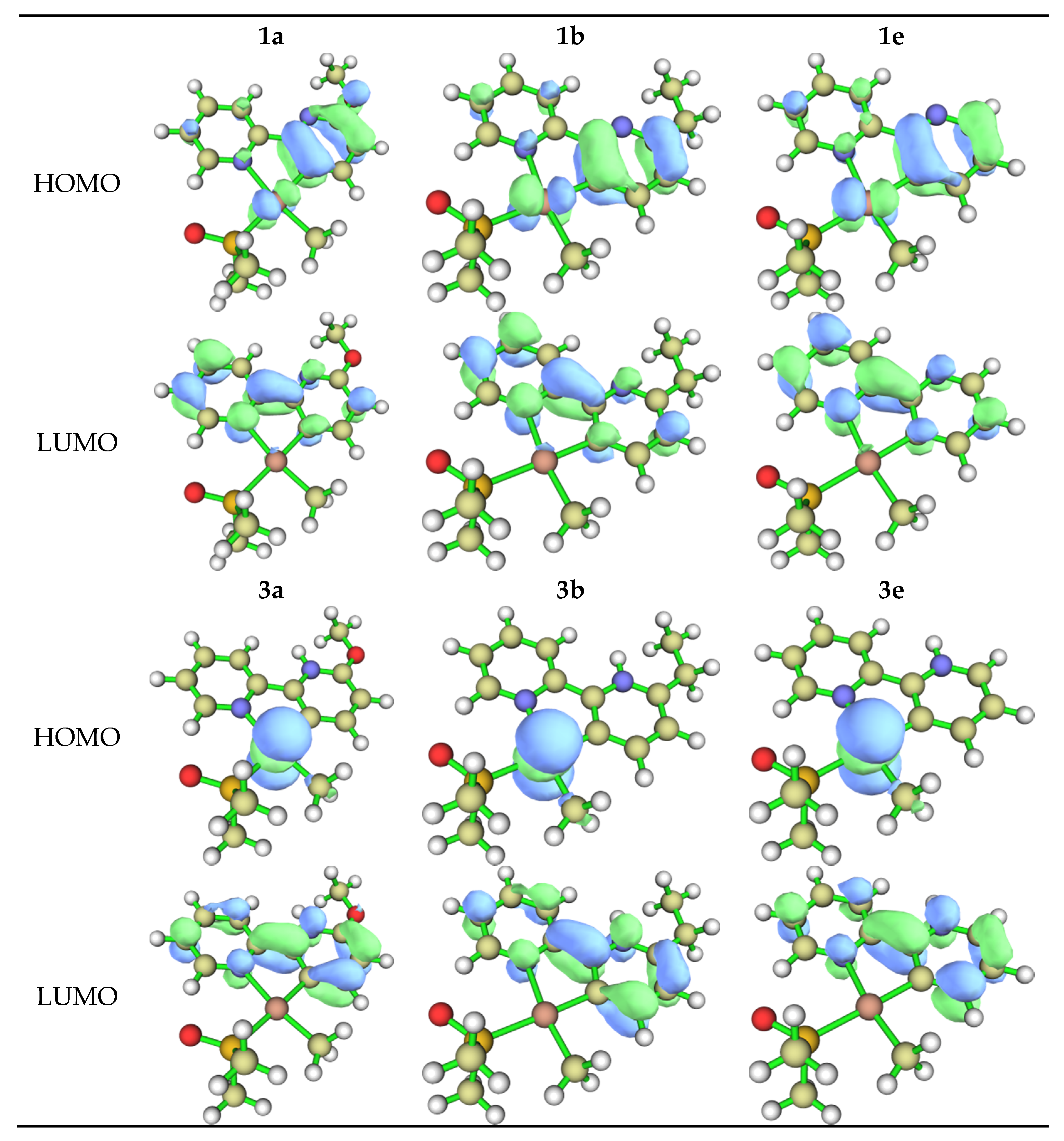
2.1. DFT Calculations
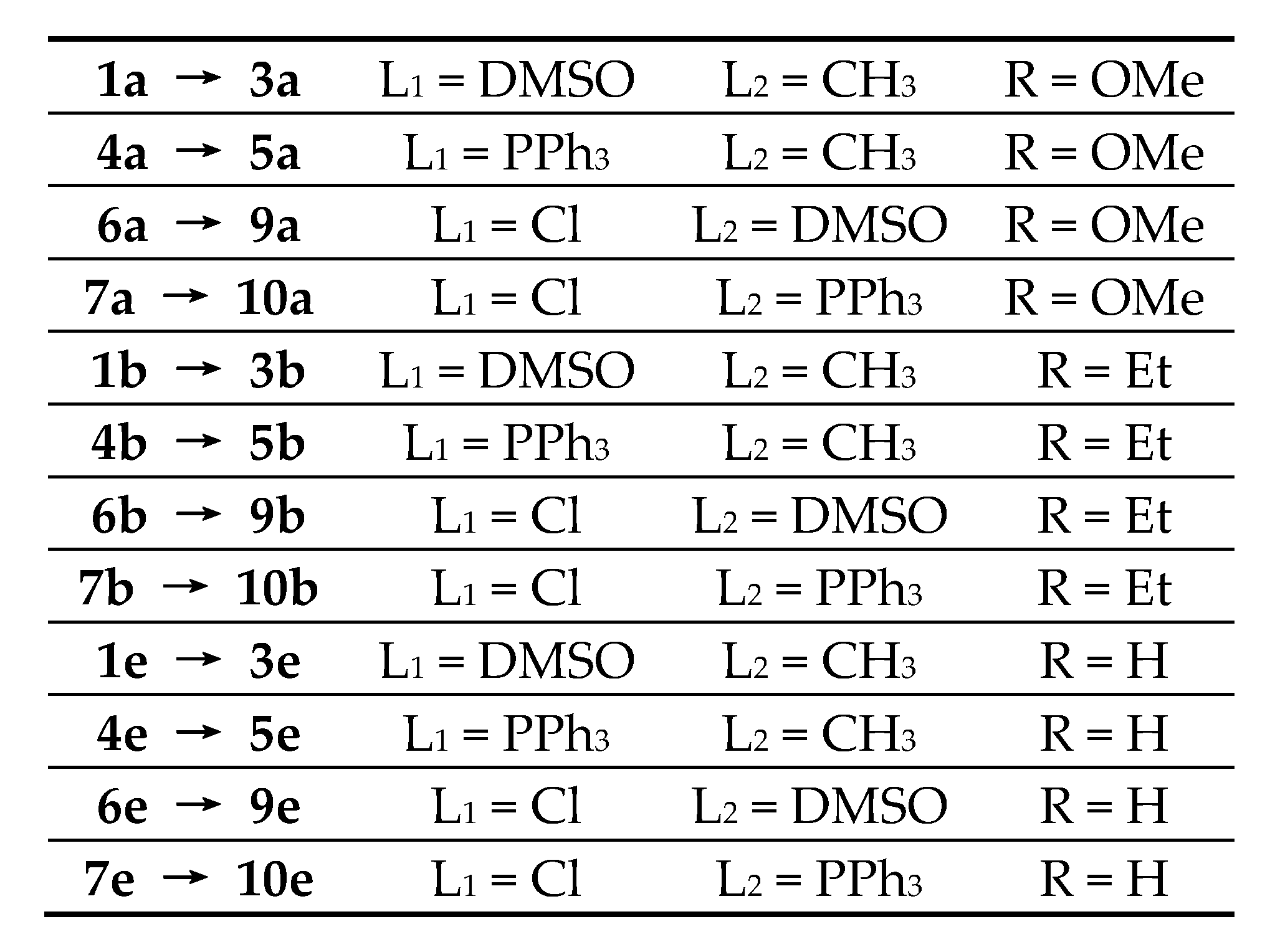
2.2. Preparations
2.3. [Pt(bpy6Et-H)(Me)(DMSO)], 1b
2.4. [.Pt(bpy6Et*)(Me)(DMSO)][BF4], 3b-BF4
2.5. [Pt(bpy6OMe-H)(Cl)(DMSO)], 4a
2.6. [Pt(bpy6Et-H)(Cl)(DMSO)], 4b
2.7. [.Pt(bpy6Et*)(Cl)(DMSO)][BF4], 5b-BF4
2.8. [Pt(bpy6Et-H)(Me)(PPh3)], 6b
2.9. Pt(bpy6OMe-H)(Cl)(PPh3)], 7a
2.10. [.Pt(bpy6Et-H)(Cl)(PPh3)], 7b
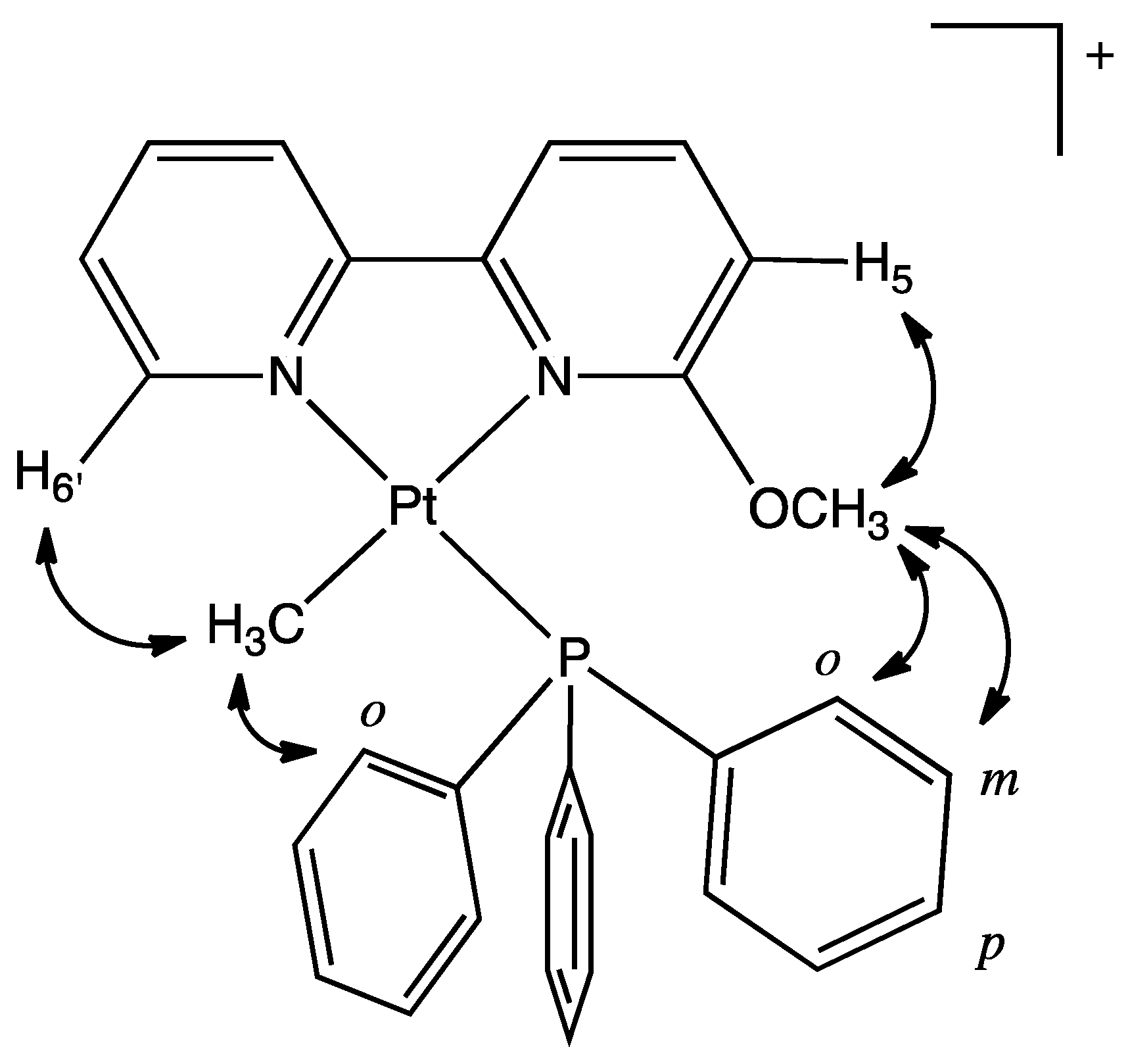
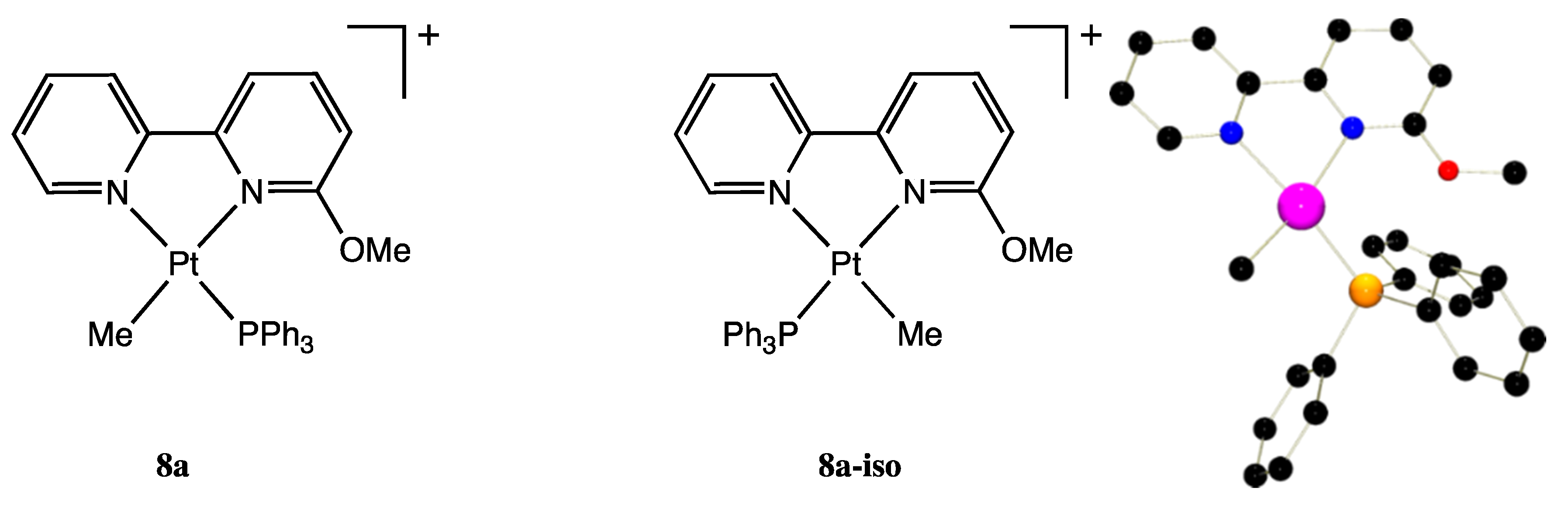
2.11. [.Pt(κ2-N,N-bpy6OMe)(Me)(PPh3)][BF4], 8a-BF4
- (a)
- To a solution of 6a (98.0 mg, 0.149 mmol) in acetone (20 mL), solid [H3O⋅18-crown-6] [BF4] was added (59.0 mg, 0.159 mmol). The solution was stirred for 30′, then it was treated with diethyl ether and filtered. Yield 85%. Anal. Calcd. for C30H28BF4N2OPPt: C, 48.34%; H, 3.79%; N, 3.76%. Found C, 47.98%; H, 3.54%; N, 4.01%.
- (b)
- To a solution of 6a (10.0 mg, 0.0152 mmol) in deuterated chloroform (2 mL) [H3O⋅18-crown-6][BF4] was added (6.5 mg, 0.017 mmol). The resulting dark yellow solution was put in an NMR tube and the reaction was followed by means of 1H, 31P, 1H COSY and 1H NOESY spectra.
2.12. [.Pt(bpy6Et*)(Me)(PPh3)][BF4], 9b-BF4
2.13. [Pt(bpy6Et*)(Cl)(PPh3)][BF4], 10b-BF4
2.14. Reaction between 1a, [Pt(bpy6OMe-H)(Me)DMSO], and [H3O⋅18-crown-6][BF4]
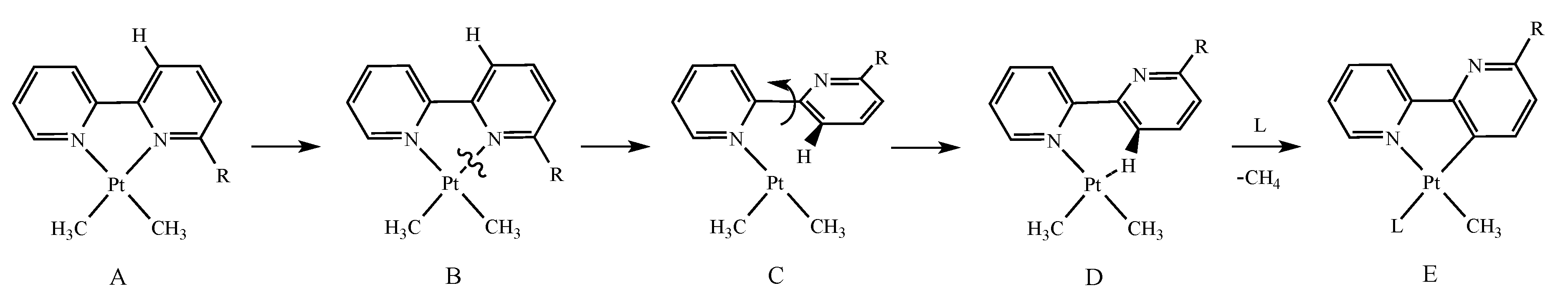
3. Results
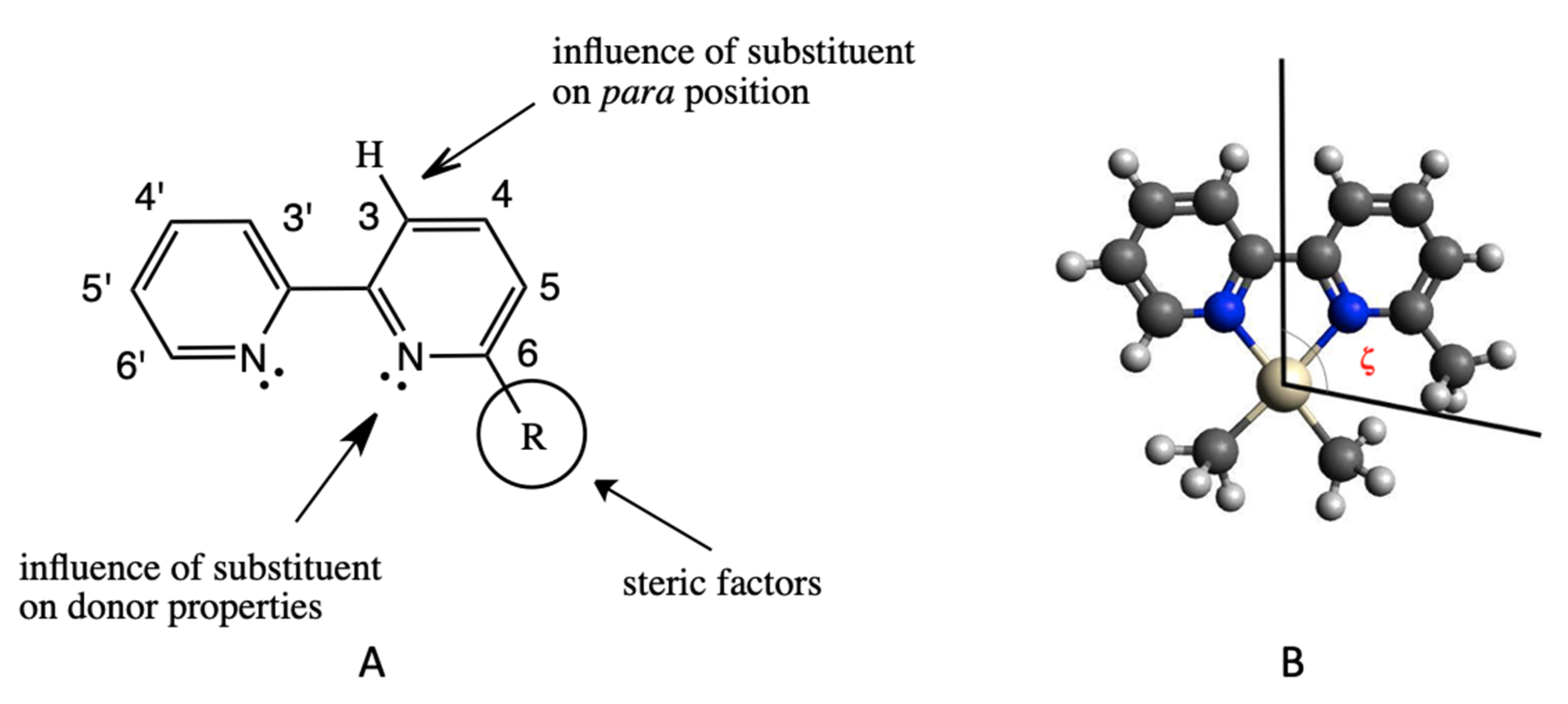
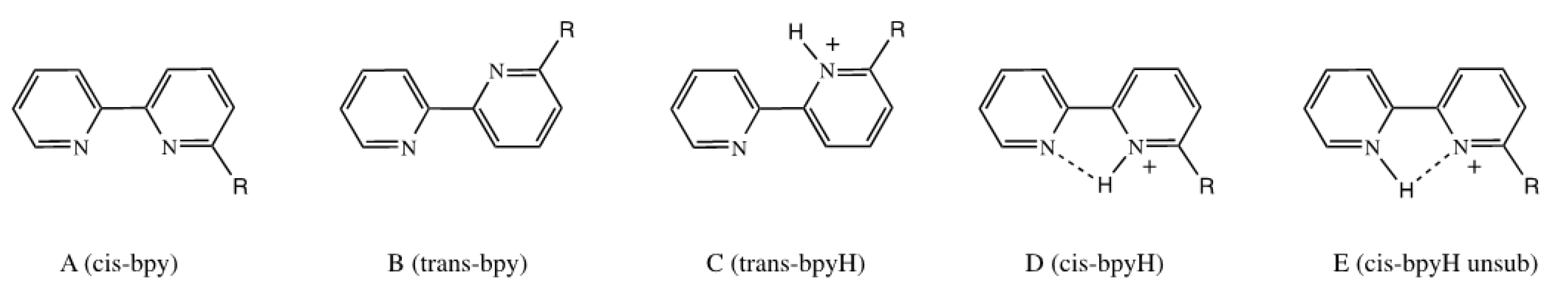
3.1. Stereoelectronic Properties of Substituted 2,2′-bipyridines
3.2. Synthesis and Characterization

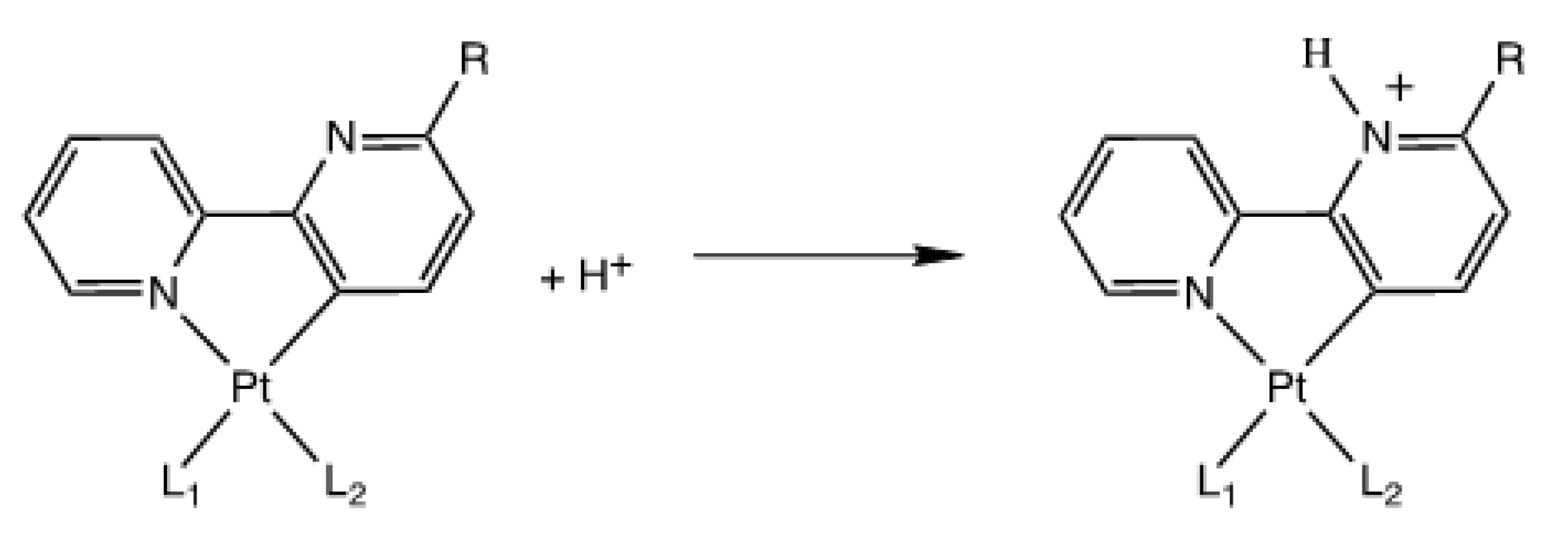
3.3. Reactivity with Acids






4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Albrecht, M. Cyclometalation using d-block transition metals: Fundamental aspects and recent trends. Chem. Rev. 2010, 110, 576–623. [Google Scholar] [CrossRef] [PubMed]
- Crespo, M. Diarylplatinum(II) Compounds as Versatile Metallating Agents in the Synthesis of Cyclometallated Platinum Compounds with N-Donor Ligands. Inorganics 2014, 2, 115–131. [Google Scholar] [CrossRef]
- Crespo, M.; Martínez, M.; Nabavizadeh, S.M.; Rashidi, M. Kinetico-mechanistic studies on C-X (X = H, F, Cl, Br, I) bond activation reactions on organoplatinum(II) complexes. Coord. Chem. Rev. 2014, 279, 115–140. [Google Scholar] [CrossRef]
- Butschke, B.; Schwarz, H. “Rollover” cyclometalation—Early history, recent developments, mechanistic insights and application aspects. Chem. Sci. 2012, 3, 308–326. [Google Scholar] [CrossRef] [Green Version]
- Zucca, A.; Petretto, G.L.; Stoccoro, S.; Cinellu, M.A.; Manassero, M.; Manassero, C.; Minghetti, G. Cyclometallation of 2,2′-bipyridine. Mono and Dinuclear C,N Platinum(II) derivatives. Organometallics 2009, 28, 2150–2159. [Google Scholar] [CrossRef]
- Aghakhanpour, R.B.; Rashidi, M.; Hosseini, F.N.; Raoofa, F.; Nabavizadeh, S.M. Oxidation of a rollover cycloplatinated(ii) dimer by MeI: A kinetic study. RSC Adv. 2015, 5, 66534–66542. [Google Scholar] [CrossRef]
- Skapski, A.C.; Sutcliffe, V.F.; Young, G.B. ‘Roll-over’ 3-Metallation of Co-ordinated 2,2’-Bipyridyl in the Thermal Rearrangement of Diaryl(bipyridyl)platinum(II) Complexes: Molecular Structure of (m-bidyl)[PtPh(Butpy)]2. J. Chem. Soc. Chem. Commun. 1985, 609–611. [Google Scholar] [CrossRef]
- Minghetti, G.; Stoccoro, S.; Cinellu, M.A.; Soro, B.; Zucca, A. Activation of a C-H Bond in a Pyridine Ring. Reaction of 6-substituted 2,2’-bipyridines with Methyl and Phenyl Platinum(II) Derivatives: N′,C(3)-“Rollover” Cyclometallation. Organometallics 2003, 22, 4770–4777. [Google Scholar] [CrossRef]
- Yang, W.; Chen, J.; Huang, X.; Ding, J.; Liu, M.; Wu, H. Pd-catalyzed intramolecular aerobic oxidative C-H amination of 2-aryl-3-(arylamino)quinazolinones: Synthesis of fluorescent indazolo[3,2-b]quinazolinones. Org. Lett. 2014, 16, 5418–5421. [Google Scholar] [CrossRef]
- Shibata, T.; Takayasu, S. Synthesis of Multicyclic Heterocycles Initiated by C–H Bond Activation Along with “Rollover” Using a Rh(III) Catalyst. Heteroatom Chem. 2014, 25, 379–388. [Google Scholar] [CrossRef]
- Yu, S.; Li, X. Mild Synthesis of Chalcones via Rhodium(III)-Catalyzed C−C Coupling of Arenes and Cyclopropenones. Org. Lett. 2014, 16, 1220–1223. [Google Scholar] [CrossRef] [PubMed]
- Kwak, J.; Ohk, Y.; Jung, Y.; Chang, S. Rollover Cyclometalation Pathway in Rhodium Catalysis: Dramatic NHC Effects in the C−H Bond Functionalization. J. Am. Chem. Soc. 2012, 134, 17778–17788. [Google Scholar] [CrossRef] [PubMed]
- Ghoochany, L.T.; Kerner, C.; Farsadpour, S.; Menges, F.; Sun, Y.; Niedner-Schatteburg, G.; Thiel, W.R. C–H Activation at a Ruthenium(II) Complex—The Key Step for a Base-Free Catalytic Transfer Hydrogenation? Eur. J. Inorg. Chem. 2013, 24, 4305–4317. [Google Scholar] [CrossRef]
- Ghorai, D.; Dutta, C.; Choudhury, J. Switching of “rollover Pathway” in Rhodium(III)-Catalyzed C-H Activation of Chelating Molecules. ACS Catal. 2016, 6, 709–713. [Google Scholar] [CrossRef]
- Zucker, S.P.; Wossidlo, F.; Weber, M.; Lentz, D.; Tzschucke, C.C. Palladium-Catalyzed Directed Halogenation of Bipyridine N-Oxide. J. Org. Chem. 2017, 82, 5616–5635. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, Y.; Wang, Z.; Feng, B.; You, J. Rhodium(III)-Catalyzed Annulation of Pyridinones with Alkynes via Double C−H Activation: A Route to Functionalized Quinolizinones. J. Org. Lett. 2017, 19, 3083–3086. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Wen, S.; Ba, D.; Lv, W.; Chen, Y.; Cheng, G. Rhodium(III)-Catalyzed Regioselective C3−H Acylmethylation of [2,2′-Bipyridine]-6-carboxamides with Sulfoxonium Ylides. Org. Lett. 2019, 21, 6366–6369. [Google Scholar] [CrossRef]
- Dutta, C.; Ghorai, D.; Choudhury, J. To “Rollover” or Not? Stereoelectronically Guided C–H Functionalization Pathways from Rhodium–Abnormal NHC Intermediates. ACS Omega 2018, 3, 1614–1620. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Lv, W.; Cheng, G. Palladium-Catalyzed Site-Selective C-H Arylation of 2,2′-Bipyridine-6-carboxamides via a Rollover Cyclometalation Pathway. Org. Lett. 2018, 20, 4732–4735. [Google Scholar] [CrossRef]
- Crabtree, R.H. Creating ligands with multiple personalities. Science 2010, 330, 455–456. [Google Scholar] [CrossRef]
- Crabtree, R.H. Abnormal, mesoionic and remote N-heterocyclic carbene complexes. Coord. Chem. Rev. 2013, 257, 755–766. [Google Scholar] [CrossRef]
- Iglesias, M.; Albrecht, M. Expanding the family of mesoionic complexes: Donor properties and catalytic impact of palladated isoxazolylidenes. Dalton Trans. 2010, 39, 5213–5215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maidich, L.; Zuri, G.; Stoccoro, S.; Cinellu, M.A.; Masia, M.; Zucca, A. Mesoionic complexes of platinum(II) derived from “rollover” cyclometalation: A delicate balance between Pt-C(sp3) and Pt-C(sp2) bond cleavage as a result of different reaction conditions. Organometallics 2013, 32, 438–448. [Google Scholar] [CrossRef]
- Schuster, O.; Yang, L.; Raubenheimer, H.G.; Albrecht, M. Beyond Conventional N-Heterocyclic Carbenes: Abnormal, Remote, and Other Classes of NHC Ligands with Reduced Heteroatom Stabilization Beyond Conventional N-Heterocyclic Carbenes: Abnormal, Remote, and Other Classes of NHC Ligands with Reduced Hetero. Chem. Rev. 2009, 109, 3445–3478. [Google Scholar] [CrossRef] [Green Version]
- Vivancos, Á.; Segarra, C.; Albrecht, M. Mesoionic and Related Less Heteroatom-Stabilized N-Heterocyclic Carbene Complexes: Synthesis, Catalysis, and Other Applications. Chem. Rev. 2018, 118, 9493–9586. [Google Scholar] [CrossRef] [PubMed]
- Leist, M.; Kerner, C.; Ghoochany, L.T.; Farsadpour, S.; Fizia, A.; Neu, J.P.; Schön, F.; Sun, Y.; Oelkers, B.; Lang, J.; et al. Roll-over cyclometalation: A versatile tool to enhance the catalytic activity of transition metal complexes. J. Organomet. Chem. 2018, 863, 30–43. [Google Scholar] [CrossRef]
- Omae, I. Unconventional Cyclometalation Reactions. Curr. Org. Chem. 2014, 18, 2776–2795. [Google Scholar] [CrossRef]
- Maidich, L.; Zucca, A.; Clarkson, G.J.; Rourke, J.P. Oxidative addition of MeI to a rollover complex of Pt(II): Isolation of the kinetic product. Organometallics 2013, 32, 3371–3375. [Google Scholar] [CrossRef]
- Maidich, L.; Zuri, G.; Stoccoro, S.; Cinellu, M.A.; Zucca, A. Assembly of symmetrical and unsymmetrical platinum(II) rollover complexes with bidentate phosphine ligands. Dalton Trans. 2014, 43, 14806–14815. [Google Scholar] [CrossRef]
- Minghetti, G.; Stoccoro, S.; Cinellu, M.A.; Petretto, G.L.; Zucca, A. “Rollover” Cyclometalated Platinum (II) Hydrides: Mono- and Polynuclear Derivatives. Organometallics 2008, 27, 3415–3421. [Google Scholar] [CrossRef]
- Hosseini, F.N.; Nabavizadeh, S.M.; Abu-omar, M.M. Which is the Stronger Nucleophile, Platinum or Nitrogen in Rollover Cycloplatinated(II) Complexes? Inorg. Chem. 2017, 56, 14706–14713. [Google Scholar] [CrossRef] [PubMed]
- Petretto, G.L.; Zucca, A.; Stoccoro, S.; Cinellu, M.A.; Minghetti, G. Step by step palladium mediated syntheses of new 2-(pyridin-2-yl)-6-R-nicotinic acids and esters. J. Organomet. Chem. 2010, 695, 256–259. [Google Scholar] [CrossRef]
- Zucca, A.; Maidich, L.; Canu, L.; Petretto, G.L.; Stoccoro, S.; Cinellu, M.A.; Clarkson, G.J.; Rourke, J.P. Rollover-assisted C(sp2)-C(sp3) bond formation. Chem. A Eur. J. 2014, 20, 5501–5510. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.Y.; Kwak, J.; Chang, S. Rhodium-catalyzed selective C–H functionalization of NNN tridentate chelating compounds via a rollover pathway. Chem. Commun. 2016, 52, 3159–3162. [Google Scholar] [CrossRef]
- Jongbloed, L.S.; De Bruin, B.; Reek, J.N.H.; Lutz, M.; Van Der Vlugt, J.I. Reversible cyclometalation at RhI as a motif for metal-ligand bifunctional bond activation and base-free formic acid dehydrogenation. Catal. Sci. Technol. 2016, 6, 1320–1327. [Google Scholar] [CrossRef] [Green Version]
- Alam, P.; Kaur, G.; Chakraborty, S.; Roy Choudhury, A.; Laskar, I.R. Aggregation induced phosphorescence active rollover iridium(III) complex as a multi-stimuli-responsive luminescence material. Dalton Trans. 2015, 44, 6581–6592. [Google Scholar] [CrossRef]
- Paziresh, S.; Babadi Aghakhanpour, R.; Rashidi, M.; Nabavizadeh, S.M. Simple tuning of the luminescence properties of the double rollover cycloplatinated(II) structure by halide ligands. New J. Chem. 2018, 42, 1337–1346. [Google Scholar] [CrossRef]
- Aghakhanpour, R.B.; Nabavizadeh, S.M.; Rashidi, M. Newly designed luminescent di- and tetra-nuclear double rollover cycloplatinated(II) complexes. J. Organomet. Chem. 2016, 819, 216–227. [Google Scholar] [CrossRef] [Green Version]
- Abedi, A.; Amani, V.; Safari, N.; Ostad, S.N.; Notash, B. From proton transferred to cyclometalated platinum(IV) complex: Crystal structure and biological activity. J. Organomet. Chem. 2015, 799–800, 30–37. [Google Scholar] [CrossRef]
- Fereidoonnezhad, M.; Shahsavari, H.R.; Abedanzadeh, S.; Behchenari, B.; Hossein-Abadi, M.; Faghih, Z.; Hassan Beyzavi, M. Cycloplatinated(II) complexes bearing 1,1′-bis(diphenylphosphino)ferrocene ligand: Biological evaluation and molecular docking studies. New J. Chem. 2018, 42, 2385–2392. [Google Scholar] [CrossRef]
- Akbarzadeh, S.; Ebrahimi, F.; Faghih, Z.; Movahed Zahra, F.A. Cytotoxic Effect Two Novel Platinum Breast Cancer: An in vitro Study. Asian Pac. J. Cancer Biol. 2018, 3, 11–14. [Google Scholar] [CrossRef] [Green Version]
- Babak, M.V.; Pfaffeneder-Kmen, M.; Meier-Menches, S.M.; Legina, M.S.; Theiner, S.; Licona, C.; Orvain, C.; Hejl, M.; Hanif, M.; Jakupec, M.A.; et al. Rollover Cyclometalated Bipyridine Platinum Complexes as Potent Anticancer Agents: Impact of the Ancillary Ligands on the Mode of Action. Inorg. Chem. 2018, 57, 2851–2864. [Google Scholar] [CrossRef] [PubMed]
- Fereidoonnezhad, M.; Niazi, M.; Shahmohammadi Beni, M.; Mohammadi, S.; Faghih, Z.; Faghih, Z.; Shahsavari, H.R. Synthesis, Biological Evaluation, and Molecular Docking Studies on the DNA Binding Interactions of Platinum(II) Rollover Complexes Containing Phosphorus Donor Ligands. ChemMedChem 2017, 12, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Giordano, T.J.; Rasmussen, P.G. Platinum and palladium complexes of thienylpyridine. Compounds containing metal-carbon bonds. Inorg. Chem. 1975, 14, 1628–1634. [Google Scholar] [CrossRef]
- Zhao, S.-B.; Wang, R.-Y.; Wang, S. Intramolecular C-H activation directed self-assembly of an organoplatinum(II) molecular square. J. Am. Chem. Soc. 2007, 129, 3092–3093. [Google Scholar] [CrossRef]
- Zucca, A.; Cinellu, M.A.; Pinna, M.V.; Stoccoro, S.; Minghetti, G.; Manassero, M.; Sansoni, M. Cyclopalladation of 6-substituted-2,2’-bipyridines. Metallation of unactivated methyl groups vs. aromatic C-H activation. Organometallics 2000, 19, 4295–4304. [Google Scholar] [CrossRef]
- Doppiu, A.; Minghetti, G.; Cinellu, M.A.; Stoccoro, S.; Zucca, A.; Manassero, M. Unprecedented Behavior of 2,2’:6’-2”-Terpyridine: Dinuclear Platinum(II) Derivatives with a new N,C; C,N Bridging ligand. Organometallics 2001, 20, 1148–1152. [Google Scholar] [CrossRef]
- Zucca, A.; Cordeschi, D.; Stoccoro, S.; Cinellu, M.A.; Minghetti, G.; Chelucci, G.; Manassero, M. Platinum(II) cyclometallated “rollover” complexes with a chiral pinene-derived 2,2′-bipyridine. Organometallics 2011, 30, 3064–3074. [Google Scholar] [CrossRef]
- Stoccoro, S.; Maidich, L.; Ruiu, T.; Cinellu, M.A.; Clarkson, G.J.; Zucca, A. Chiral cyclometalation of 6-(1-phenylbenzyl)-2,2′-bipyridine. Dalton Trans. 2015, 44, 18001–18011. [Google Scholar] [CrossRef] [Green Version]
- Cocco, F.; Zucca, A.; Stoccoro, S.; Guerri, A.; Serratrice, M.; Cinellu, M.A. Synthesis and Characterization of Palladium(II) and Platinum(II) Adducts and Cyclometalated Complexes of 6,6′-Dimethoxy-2,2′-bipyridine—C(sp3)-H and C(sp2)-H Bond Activations. Organometallics 2014, 33, 3414–3424. [Google Scholar] [CrossRef]
- Vogel, A.I. Vogel’s Textbook of Practical Organic Chemistry, 5th ed.; Longman Scientific and Technical: Harlow, UK, 1989. [Google Scholar]
- Eaborn, C.; Kundu, K.; Pidcock, A. Synthesis of platinum(II) alkyl and aryl complexes from K2[PtCl4] and tetraorganotin compounds in dimethyl sulphoxide. J. Chem. Soc. Dalton Trans. 1981, 933–938. [Google Scholar] [CrossRef]
- Romeo, R.; Monsu Scolaro, L. (2,2:6,2″-terpyridine)methylplatinum(II) chloride and (1,10-phenanthroline) methylchloroplatinum(II). Inorg. Synth. 1998, 32, 153. [Google Scholar]
- Baquero, E.A.; Rodríguez-Zúñiga, A.; Flores, J.C.; Temprado, M.; de Jesús, E. Revisiting the synthesis of trans-[Pt(dmso)2ClMe] and cis-[Pt(dmso)2Me2]: Experimental and DFT studies. J. Organomet. Chem. 2019, 896, 108–112. [Google Scholar] [CrossRef]
- Gütz, C.; Lützen, A. Synthesis of 2,2′-Bipyridines via Suzuki-Miyaura Cross-Coupling. Synthesis 2010, 85–90. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Erratum Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef] [Green Version]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic regular two-component Hamiltonians. J. Chem. Phys. 1993, 99, 4597–4610. [Google Scholar] [CrossRef]
- Chang, C.; Pelissier, M.; Durand, P. Regular Two-Component Pauli-Like Effective Hamiltonians in Dirac Theory. Phys. Scr. 1986, 34, 394–404. [Google Scholar] [CrossRef]
- Heully, J.L.; Lindgren, I.; Lindroth, E.; Lundqvist, S.; Martensson-Pendrill, A.M. Diagonalisation of the Dirac Hamiltonian as a basis for a relativistic many-body procedure. J. Phys. B At. Mol. Phys. 1986, 19, 2799–2815. [Google Scholar] [CrossRef]
- van Lenthe, E.; Ehlers, A.; Baerends, E.-J. Geometry optimizations in the zero order regular approximation for relativistic effects. J. Chem. Phys. 1999, 110, 8943–8953. [Google Scholar] [CrossRef] [Green Version]
- Pantazis, D.A.; Chen, X.-Y.; Landis, C.R.; Neese, F. All-Electron Scalar Relativistic Basis Sets for Third-Row Transition Metal Atoms. J. Chem. Theory Comput. 2008, 4, 908–919. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. WIREs. Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Maidich, L.; Cinellu, M.A.; Cocco, F.; Stoccoro, S.; Sedda, M.; Galli, S.; Zucca, A. Platinum(II), palladium(II) and gold(III) adducts and cyclometalated derivatives of 6-methoxy-2,2’-bipyridine: A comparative study. J. Organomet. Chem. 2016, 819, 76–86. [Google Scholar] [CrossRef]
- Maidich, L.; Dettori, G.; Stoccoro, S.; Cinellu, M.A.; Rourke, J.P.; Zucca, A. Electronic and steric effects in rollover C-H bond activation. Organometallics 2015, 34, 817–828. [Google Scholar] [CrossRef]
- Aue, D.H.; Webb, H.M.; Davidson, W.R.; Toure, P.; Hopkins, H.P.; Moulik, S.P.; Jahagirdar, D.V.; Hopkins, H.P.; Moulik, S.P.; Jahagirdar, D.V. Relationships between the Thermodynamics of Protonation in the Gas and Aqueous Phase for 2-, 3-, and 4-Substituted Pyridines (Proton affinity and basicity data). J. Am. Chem. Soc. 1991, 113, 1770–1780. [Google Scholar] [CrossRef]
- pKa Data of Pyridinium Ions, Compiled by Williams, R. Available online: http://research.chem.psu.edu/brpgroup/pKa_compilation.pdf (accessed on 24 August 2020).
- Hunter, E.P.L.; Lias, S.G. Evaluated Gas Phase Basicities and Proton Affinities of Molecules: An Update. J. Phys. Chem. Ref. Data 1998, 27, 413–656. [Google Scholar] [CrossRef]
- McMurry, J. Organic Chemistry, 8th ed.; Brooks/Cole, Cengage Learning: Belmont, CA, USA, 2012; p. 584. [Google Scholar]
- Zucca, A.; Doppiu, A.; Cinellu, M.A.; Minghetti, G.; Stoccoro Manassero, M. Multiple C-H bond activation. 3-fold deprotonated 6-phenyl-2,2′-bipyridine as a bridging ligand in dinuclear platinum(II) derivatives. Organometallics 2002, 21, 783–785. [Google Scholar] [CrossRef]
- Zucca, A.; Stoccoro, S.; Cinellu, M.A.; Minghetti, G.; Manassero, M.; Sansoni, M. Metallation of unactivated methyl groups. Platinum(II) derivatives with 6-alkyl-2,2’-bipyridines. Eur. J. Inorg. Chem. 2002, 3336–3346. [Google Scholar] [CrossRef]
- Zucca, A.; Maidich, L.; Carta, V.; Petretto, G.L.; Stoccoro, S.; Cinellu, M.A.; Pilo, M.I.; Clarkson, G.J. Cyclometalated Complexes of Platinum(II) with 2-Vinylpyridine. Eur. J. Inorg. Chem. 2014, 2014, 2278–2287. [Google Scholar] [CrossRef]
- Chassot, L.; Mueller, E.; von Zelewsky, A. cis-Bis(2-phenylpyridine)platinum(II) (CBPPP): A simple molecular platinum compound. Inorg. Chem. 1984, 23, 4249–4253. [Google Scholar] [CrossRef]
- Zucca, A.; Cordeschi, D.; Maidich, L.; Pilo, M.I.; Masolo, E.; Stoccoro, S.; Cinellu, M.A.; Galli, S. Rollover cyclometalation with 2-(2′-pyridyl)quinoline. Inorg. Chem. 2013, 52, 7717–7731. [Google Scholar] [CrossRef] [PubMed]
- Hallett, A.J.; Kariuki, B.M.; Pope, S.J.A. New 2,3-disubstituted-5-hydroxyquinoxaline ligands and their coordination chemistry with cyclometallated iridium(III): Syntheses, structures and tunable electronic properties. Dalton Trans. 2011, 40, 9474–9481. [Google Scholar] [CrossRef]
- Jamali, S.; Nabavizadeh, S.M.; Rashidi, M. Oxidative Addition of Methyl Iodide to a New Type of Binuclear Platinum(II) Complex: A Kinetic Study. Inorg. Chem. 2005, 44, 8594–8601. [Google Scholar] [CrossRef]
- Aghakhanpour, R.B.; Nabavizadeh, S.M.; Mohammadi, L.; Jahromi, S.A.; Rashidi, M. A kinetic approach to carbon–iodide bond activation by rollover cycloplatinated(II) complexes containing monodentate phosphine ligands. J. Organomet. Chem. 2015, 781, 47–52. [Google Scholar] [CrossRef]
- Crabtree, R.H. The Organometallic Chemistry of the Transition Metals, 6th ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2018; pp. 155–156. [Google Scholar]
- Maksić, Z.B.; Kovačević, B.; Vianello, R. Advances in determining the absolute proton affinities of neutral organic molecules in the gas phase and their interpretation: A theoretical account. Chem. Rev. 2012, 112, 5240–5270. [Google Scholar] [CrossRef]
- Grummt, U.W.; Erhardt, S. Torsional profiles of protonated and metal-coordinated 2,20-bipyridine. J. Mol. Struct. 2004, 685, 133–137. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
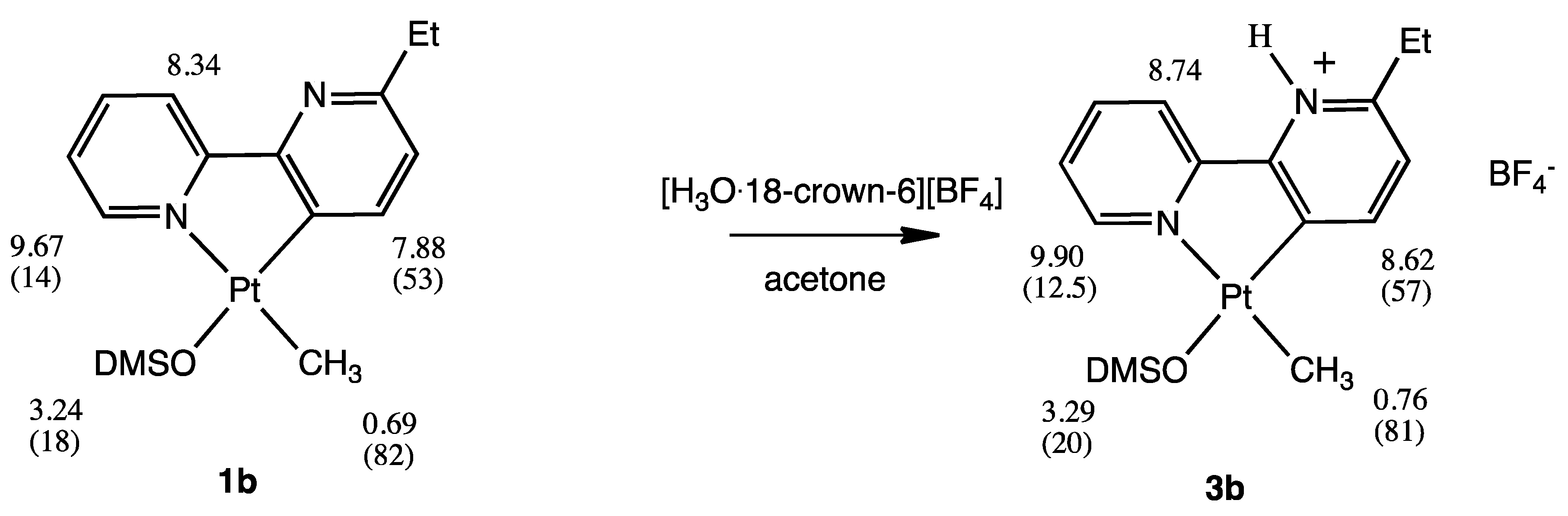{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| R a | Proton Affinity 69 | Basicity 69 | pKa Pyridinium 70 | ζ Angle (°) |
|---|---|---|---|---|
| H | 922.2 | 889.5 | 5.17 | 98.8 |
| 2-CH3 (6-CH3) | 936.0 | 903.3 | 5.97 | 125.1 |
| 3-CH3 (5-CH3) | 932.2 | 899.6 | 5.68 | |
| 4-CH3 | 936.0 | 903.3 | 6.02 | |
| 2-C2H5 (6-C2H5) | 941.0 | 908.3 | 5.97 | 157.1 (max), 125.1 (min) |
| 3-C2H5 (6-C2H5) | 936.8 | 904.2 | 5.70 | |
| 4-C2H5 | 939.7 | 907.1 | 6.02 | |
| 2-CF3 (6-CF3) | 885.3 | 852.7 | - | 137.6 |
| 3-CF3 (6-CF3) | 890.4 | 857.7 | 3.36 71 | |
| 4-CF3 | 891.6 | 859.0 | 3.59 71 | |
| 2-OCH3 (6-OCH3) | 925.9 | 893.3 | 3.28 | 152.3 (max), 109.3 (min) |
| 3-OCH3 (6-OCH3) | 930.9 | 898.3 | 4.88 | |
| 4-OCH3 | 948.9 | 916.3 | 6.62 |
| CH3 | DMSO | H6′ | H4 | ||
|---|---|---|---|---|---|
| [Pt(bpy6OMe-H)(Me)(DMSO)] | 1a | 0.69 (82) | 3.24 (18) | 9.65 (15) | 7.88 (50) |
| [Pt(bpy6Et-H)(Me)(DMSO)] | 1b | 0.69 (82) | 3.24 (18) | 9.67 (13.5) | 7.92 (53) |
| [Pt(bpy6Me-H)(Me)(DMSO)] | 1c | 0.69 (82) | 3.24 (18.5) | 9.67 (14) | 7.88 (53) |
| [Pt(bpy6CF3-H)(Me)(DMSO)] | 1d | 0.74 (82) | 3.26 (18.5) | 9.73 (14) | 8.15 (56) |
| [Pt(bpy-H)(Me)(DMSO)] | 1e | 0.70 (82) | 3.25 (18.3) | 9.71 (14) | 8.01 (54) |
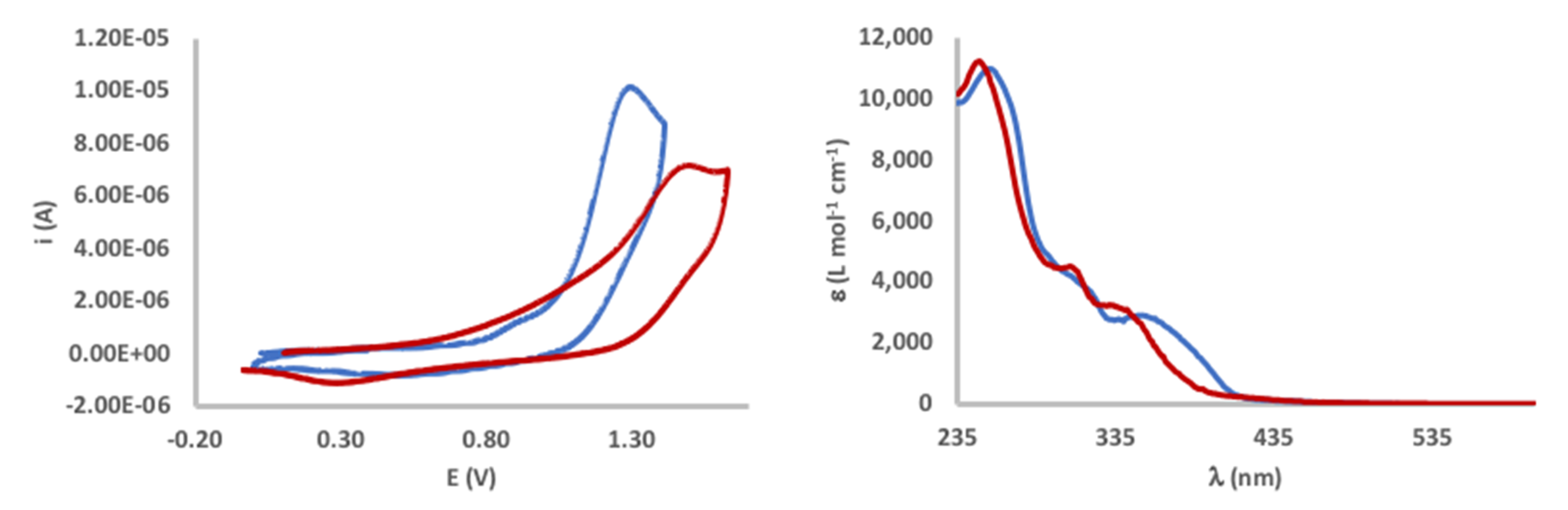
| λ (nm) a | Eg,opt (eV) b | Eox (V) c | HOMO (eV) d | LUMO (eV) e | |
|---|---|---|---|---|---|
| [Pt(bpyOMe-H)(Me)(DMSO)] 1a | 255, 352 | 3.03 | 1.29 | −5.59 | −2.56 |
| [Pt(bpyEt-H)(Me)(DMSO)] 1b | 249, 309, 340 | 3.14 | 1.4–1.5 (broad) | −5.4 ca. f | −2.3 ca. f |
| [Pt(bpy-H)(Me)(DMSO)] 1e | 276, 307, 380 | 2.99 | 0.72 | −5.13 | −2.14 |
| Neutral/Protonated | Ancillary Ligands | Bpy6ome | Bpy6et | Bpy | |
| Free ligands | bpyR - bpyRH+ (a) B → C | 952.22 | 968.30 | 948.51 | |
| bpyR - bpyRH+ (b) B → D (E) | 985.05 (E) (c) | 1002.76 | 982.72 | ||
| complexes | 1–3 | Me/DMSO | 970.85 | 1005.23 | 990.15 |
| 4–5 | Cl/DMSO | 954.78 | 988.48 | 972.45 | |
| 6–9 | Me/PPh3 | 992.85 | 1027.63 | 1014.17 | |
| 7–10 | Cl/PPh3 | 972.22 | 1006.37 | 992.51 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zucca, A.; Maidich, L.; Pilo, M.I.; Pischedda, S.; Sedda, M.; Stoccoro, S. Pt(II) Derivatives with Rollover-Coordinated 6-substituted 2,2′-bipyridines: Ligands with Multiple Personalities. Appl. Sci. 2020, 10, 6665. https://doi.org/10.3390/app10196665
Zucca A, Maidich L, Pilo MI, Pischedda S, Sedda M, Stoccoro S. Pt(II) Derivatives with Rollover-Coordinated 6-substituted 2,2′-bipyridines: Ligands with Multiple Personalities. Applied Sciences. 2020; 10(19):6665. https://doi.org/10.3390/app10196665
Chicago/Turabian StyleZucca, Antonio, Luca Maidich, Maria I. Pilo, Sara Pischedda, Mondina Sedda, and Sergio Stoccoro. 2020. "Pt(II) Derivatives with Rollover-Coordinated 6-substituted 2,2′-bipyridines: Ligands with Multiple Personalities" Applied Sciences 10, no. 19: 6665. https://doi.org/10.3390/app10196665