Dietary α-Linolenic Acid Counters Cardioprotective Dysfunction in Diabetic Mice: Unconventional PUFA Protection

 , , ,
, , ,
Abstract
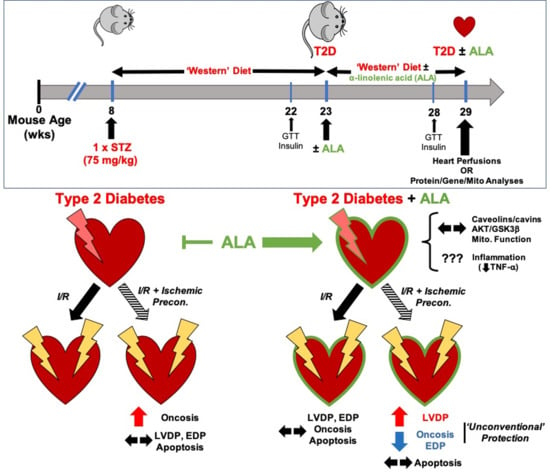
:
1. Introduction
2. Materials and Methods
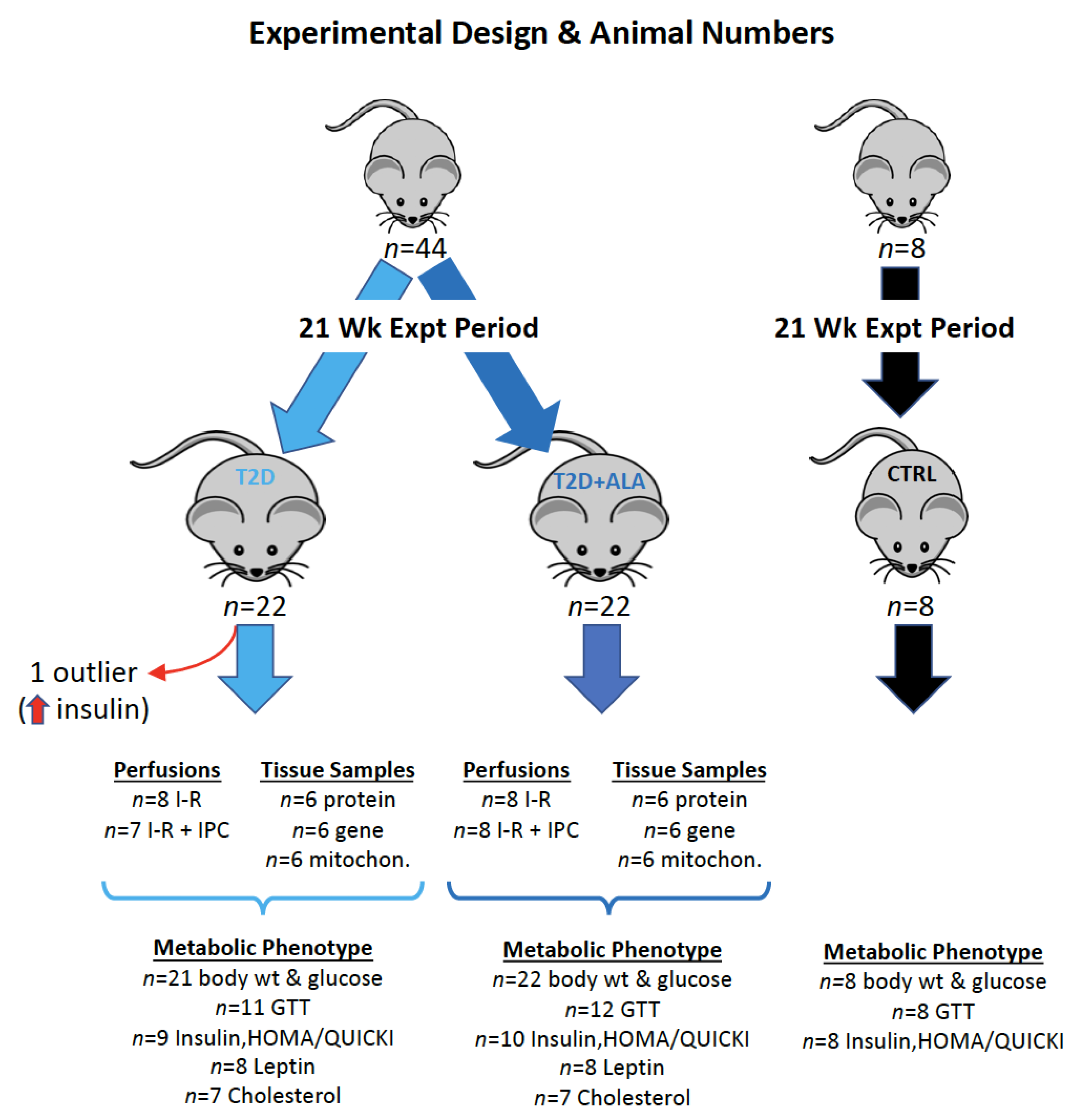
2.1. Experimental Animals and Ethics
2.2. Murine T2D Model and ALA Supplementation
2.3. Assessment of Metabolic Phenotype
2.4. Heart Perfusion and Responses to I-R ± IPC
2.5. Myocardial Tissue Fractionation and Protein Analysis
2.6. Mitochondrial Respiratory Function
2.7. Myocardial and Hepatic Inflammatory Mediator Profiles
2.8. Statistical Analyses
3. Results
3.1. Metabolic Phenotype
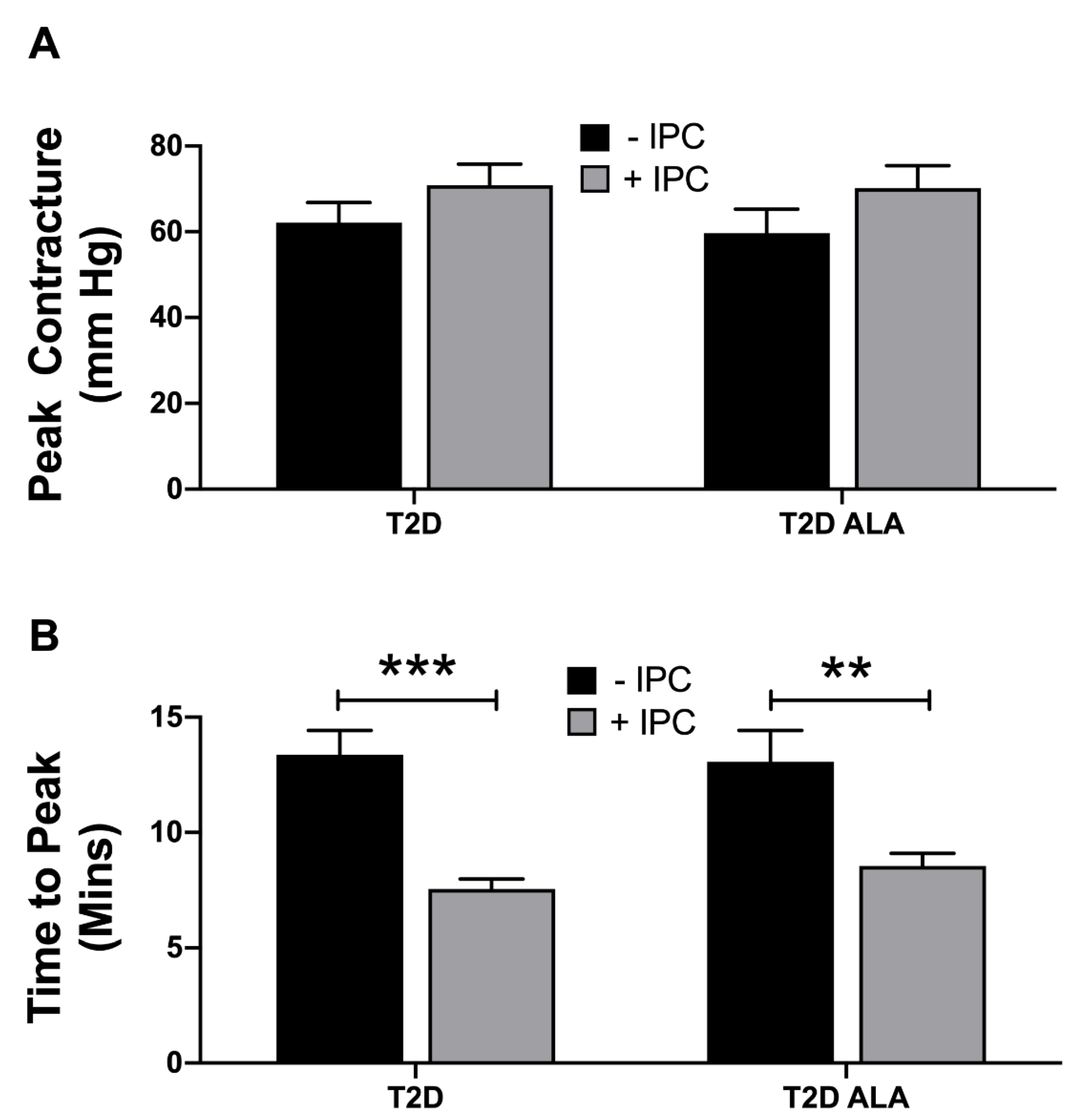
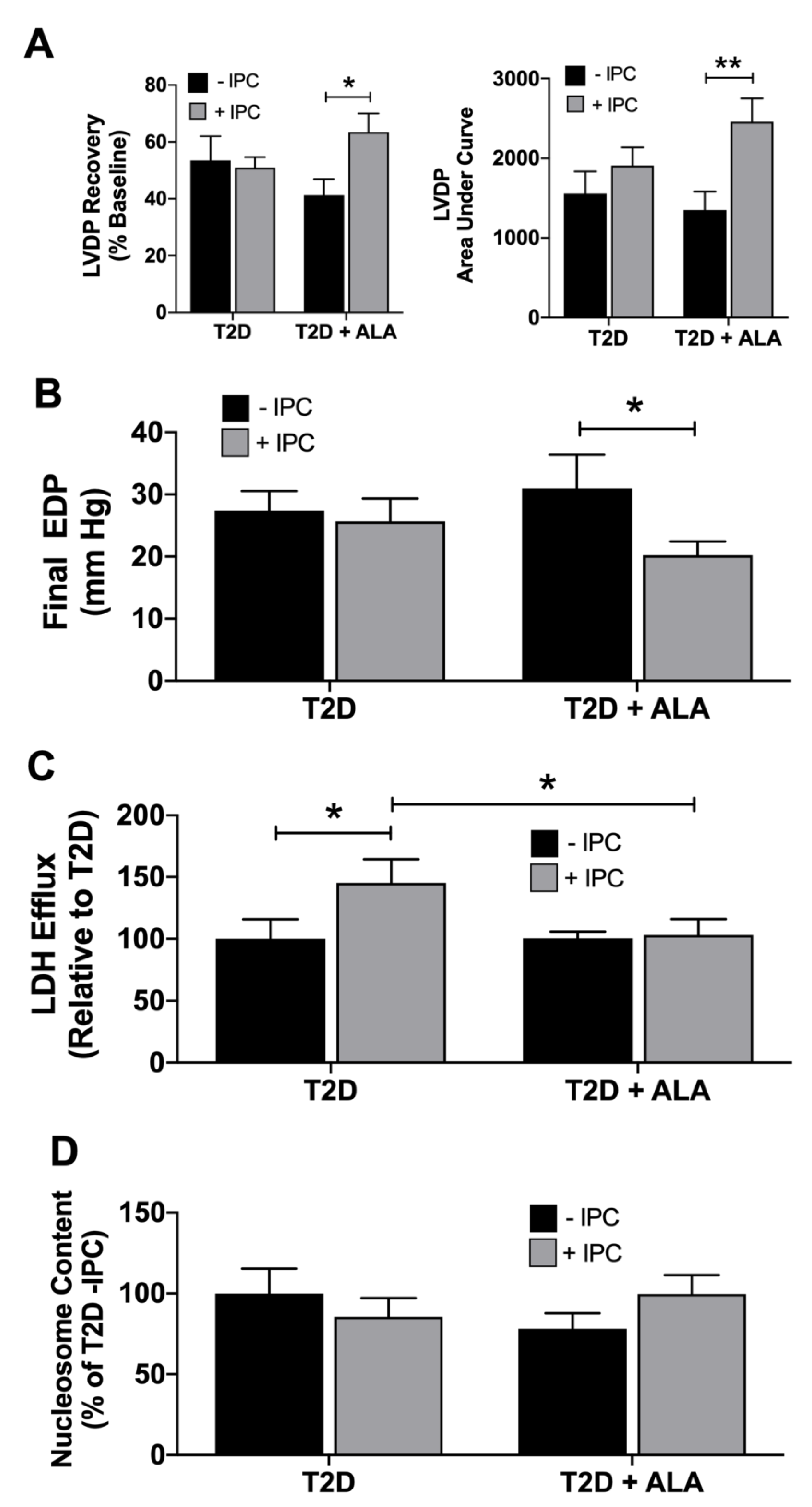
3.2. Myocardial Function, I-R Tolerance and Responses to IPC
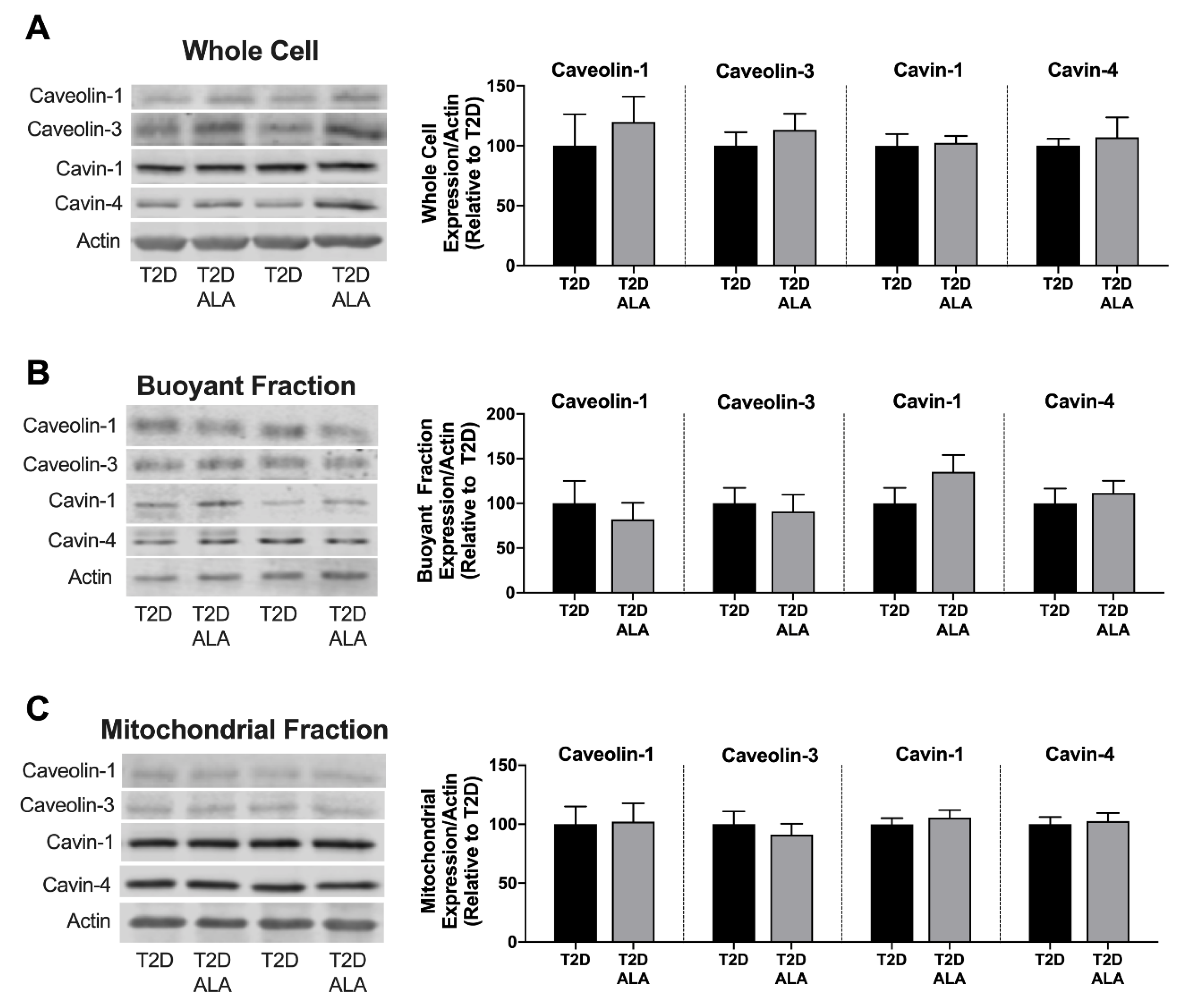
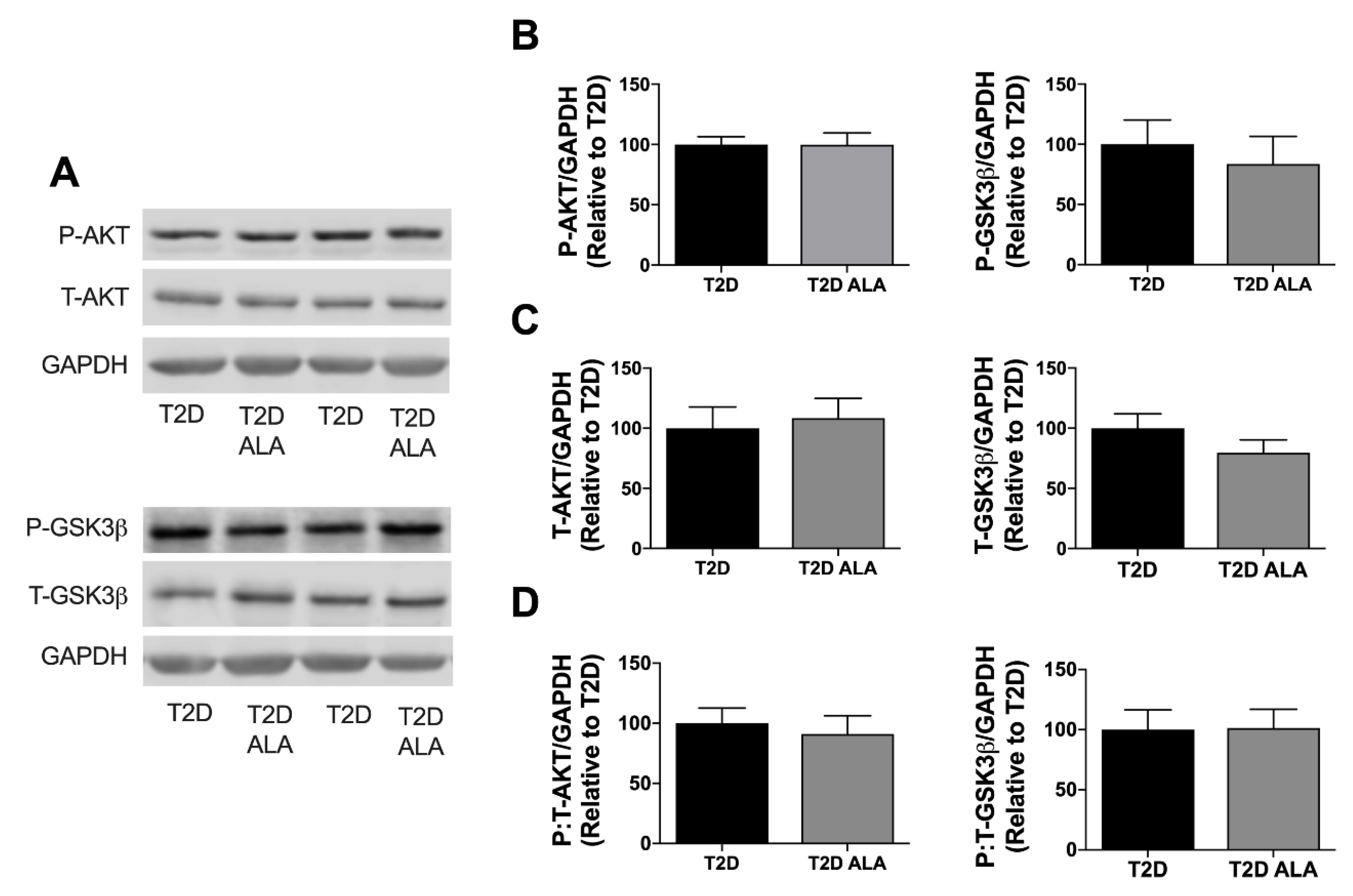
3.3. Cardiac Caveolar Proteins and Survival Kinases
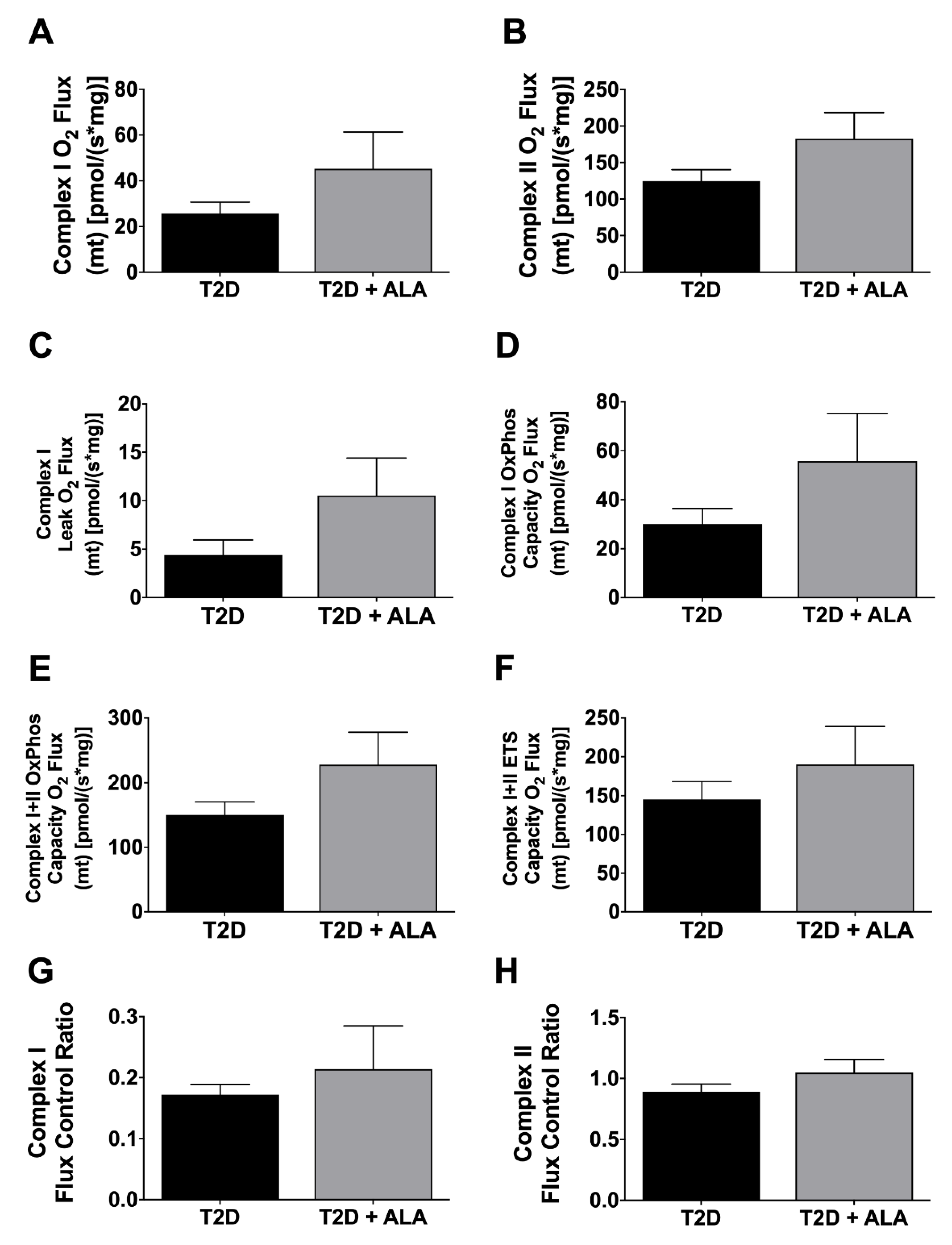
3.4. Cardiac Mitochondrial Function
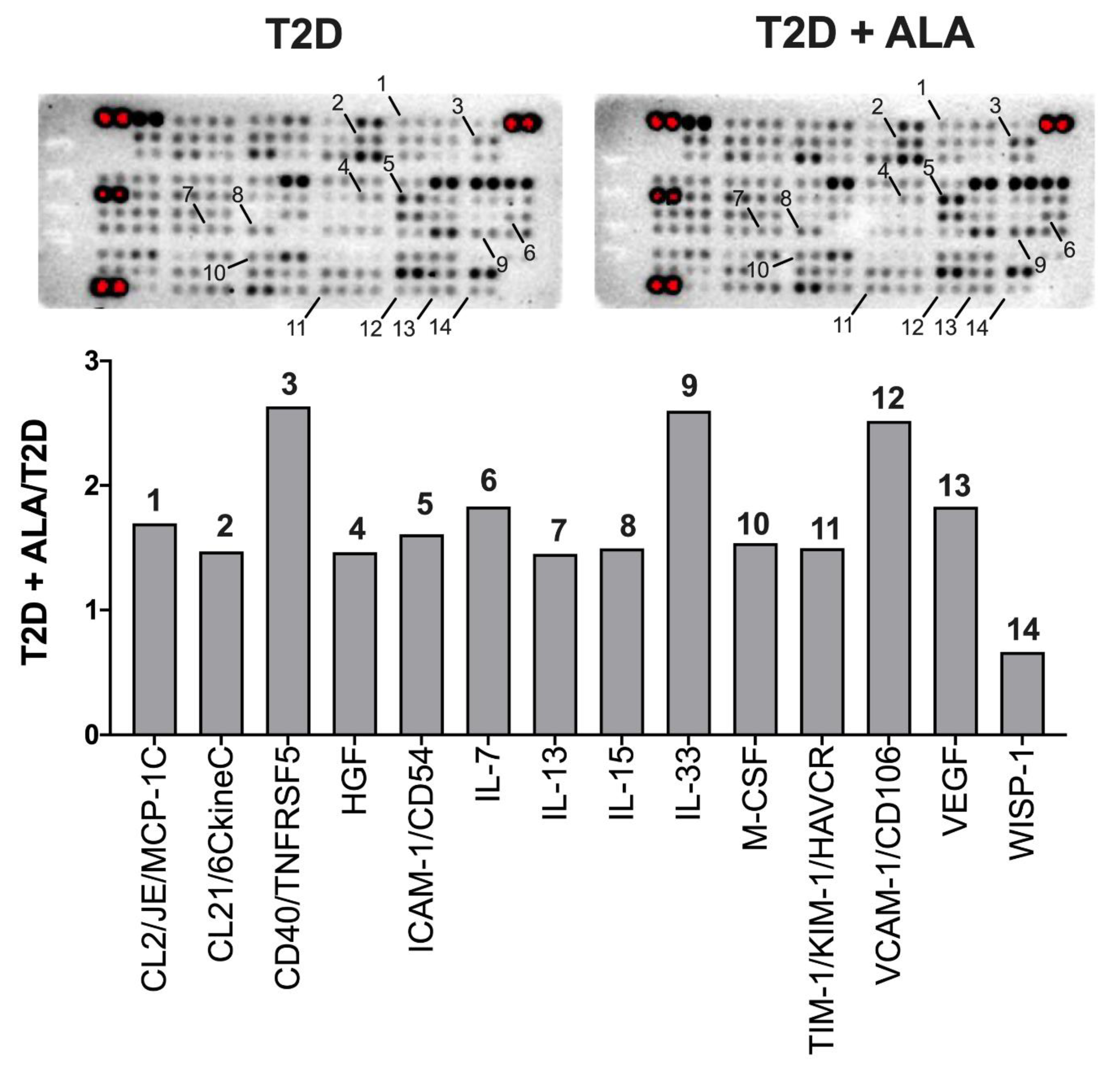
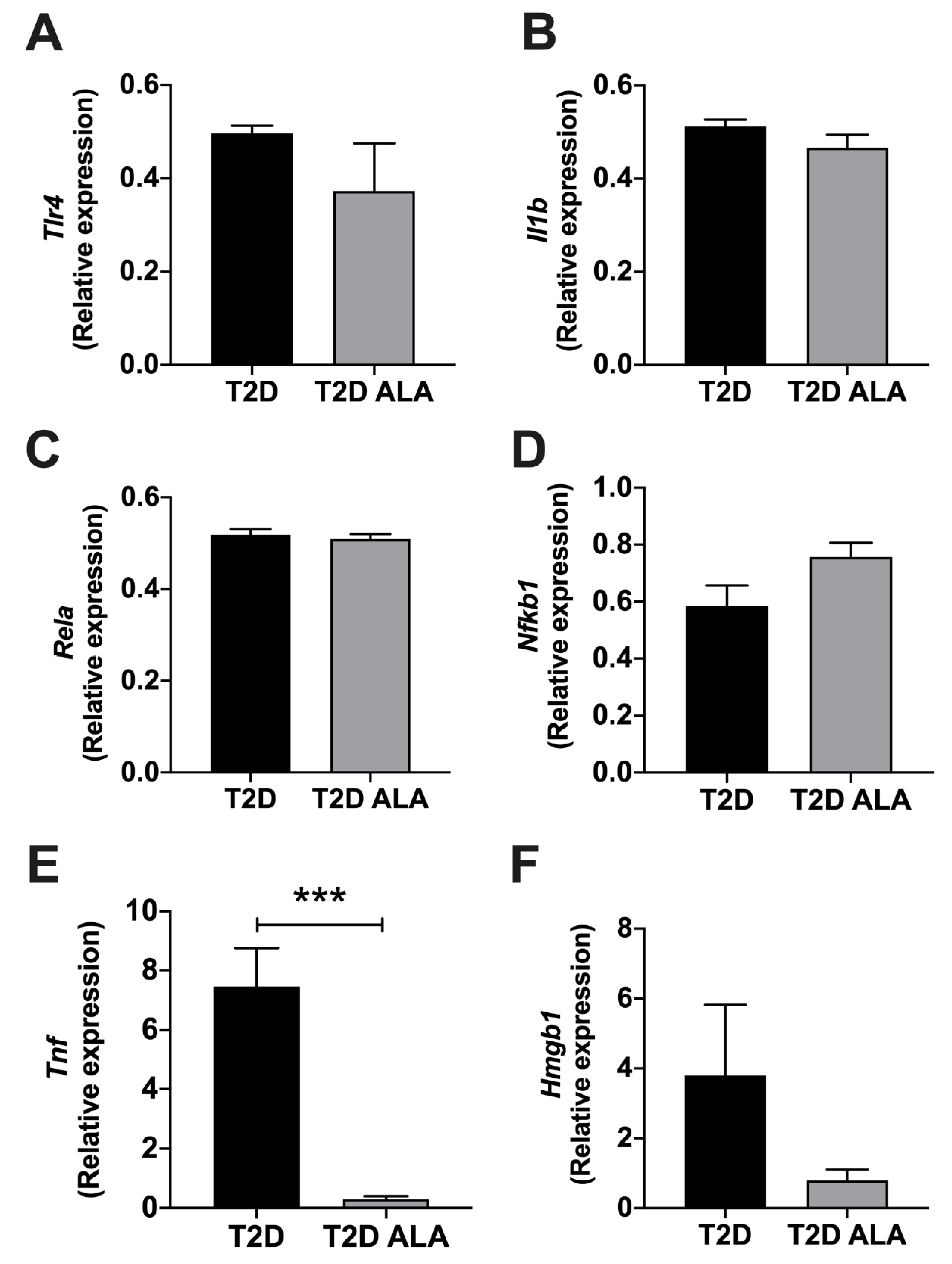
3.5. Myocardial Inflammatory Mediators
4. Discussion
4.1. Systemic Phenotype
4.2. Myocardial Function, Ischemic Tolerance and Preconditioning
4.3. Caveolar Protein Expression and Localization
4.4. Myocardial Kinase Expression and Phosphorylation
4.5. Mitochondrial Function
4.6. Inflammatory Mediators
4.7. Limitations and Future Studies
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

- GTTs were performed in 12 mice from each group (giving n = 11 for T2D after outlier removal, and n = 12 for T2D + ALA).
- Fasting insulin, HOMA and QUICKI were determined in 10 mice per group, giving n = 9 for T2D (after outlier removal), and n = 10 for T2D + ALA.
- Sufficient serum remained for analysis of Leptin in n = 8 mice/group, and cholesterol in n = 7 mice/group.
Appendix B

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | 14 Weeks | 20 Weeks | n/Group |
|---|---|---|---|
| CTRL | 9.0 ± 0.8 | 8.1 ± 0.6 | 8 |
| T2D | 11.3 ± 0.5 * | 11.8 ± 0.4 * | 21 |
| T2D + ALA | 11.2 ± 0.4 * | 12.0 ± 0.3 * | 22 |
Appendix C
| Target | Primer Sequence | Product Size (bp) | Reference |
|---|---|---|---|
| Tlr4 | F-ACCTGGCTGGTTTACACGTC R-CTGCCAGAGACATTGCAGAA | 201 | [139] |
| Il1b | F-ATGAGAGCATCCAGCTTCAA R-TGAAGGAAAAGAAGGTGCTC | 156 | [140] |
| Rela | F-CTTCCTCAGCCATGGTACCTCT R-CAAGTCTTCATCAGCATCAAACTG | 167 | [141] |
| Nfkb1 | F-GAAATTCCTGATCCAGACAAAAAC R-ATCACTTCAATGGCCTCTGTGTAG | 194 | [141] |
| Tnf | F-GCCTCTTCTCATTCCTGCTTG R-CTGATGAGAGGGAGGCCATT | 115 | [142] |
| Hmgb1 | F-TGGCAAAGGCTGACAAGGCTC R-GGATGCTCGCCTTTGATTTTGG | 166 | [143] |
Appendix D
| EDP (mmHg) | SysP (mmHg) | LVDP (mmHg) | +dP/dt (mmHg/s) | −dP/dt (mmHg/s) | Coronary Flow (mL/min) | |
|---|---|---|---|---|---|---|
| T2D (n = 16) | 5 ± 1 | 121 ± 4 | 116 ± 4 | 4340 ± 167 | −3190 ± 197 | 4.9 ± 0.8 |
| T2D + ALA (n = 16) | 5 ± 1 | 117 ± 5 | 112 ± 5 | 4202 ± 198 | −2539 ± 201 * | 4.3 ± 0.4 |
References
- Mozaffarian, D.; Wu, J.H. Omega-3 fatty acids and cardiovascular disease: Effects on risk factors, molecular pathways, and clinical events. J. Am. Coll. Cardiol. 2011, 58, 2047–2067. [Google Scholar] [CrossRef] [Green Version]
- Sokola-Wysoczanska, E.; Wysoczanski, T.; Wagner, J.; Czyz, K.; Bodkowski, R.; Lochynski, S.; Patkowska-Sokola, B. Polyunsaturated Fatty Acids and Their Potential Therapeutic Role in Cardiovascular System Disorders—A Review. Nutrients 2018, 10, 1561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelhamid, A.S.; Brown, T.J.; Brainard, J.S.; Biswas, P.; Thorpe, G.C.; Moore, H.J.; Deane, K.H.; AlAbdulghafoor, F.K.; Summerbell, C.D.; Worthington, H.V.; et al. Omega-3 fatty acids for the primary and secondary prevention of cardiovascular disease. Cochrane Database Syst. Rev. 2018, 11, CD003177. [Google Scholar] [CrossRef] [PubMed]
- Brown, T.J.; Brainard, J.; Song, F.; Wang, X.; Abdelhamid, A.; Hooper, L.; Group, P. Omega-3, omega-6, and total dietary polyunsaturated fat for prevention and treatment of type 2 diabetes mellitus: Systematic review and meta-analysis of randomised controlled trials. BMJ 2019, 366, l4697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.B.; Niaz, M.A.; Sharma, J.P.; Kumar, R.; Rastogi, V.; Moshiri, M. Randomized, double-blind, placebo-controlled trial of fish oil and mustard oil in patients with suspected acute myocardial infarction: The Indian experiment of infarct survival-4. Cardiovasc. Drugs Ther. 1997, 11, 485–491. [Google Scholar] [CrossRef]
- Folino, A.; Sprio, A.E.; Di Scipio, F.; Berta, G.N.; Rastaldo, R. Alpha-linolenic acid protects against cardiac injury and remodelling induced by beta-adrenergic overstimulation. Food Funct. 2015, 6, 2231–2239. [Google Scholar] [CrossRef]
- Ganguly, R.; Hasanally, D.; Stamenkovic, A.; Maddaford, T.G.; Chaudhary, R.; Pierce, G.N.; Ravandi, A. Alpha linolenic acid decreases apoptosis and oxidized phospholipids in cardiomyocytes during ischemia/reperfusion. Mol. Cell. Biochem. 2018, 437, 163–175. [Google Scholar] [CrossRef]
- Gao, K.; Chen, L.; Yang, M.; Han, L.; Yiguang, S.; Zhao, H.; Chen, X.; Hu, W.; Liang, H.; Luo, J.; et al. Marine n-3 PUFA protects hearts from I/R injury via restoration of mitochondrial function. Scand. Cardiovasc. J. 2015, 49, 264–269. [Google Scholar] [CrossRef]
- Demaison, L.; Moreau, D.; Vergely-Vandriesse, C.; Gregoire, S.; Degois, M.; Rochette, L. Effects of dietary polyunsaturated fatty acids and hepatic steatosis on the functioning of isolated working rat heart under normoxic conditions and during post-ischemic reperfusion. Mol. Cell. Biochem. 2001, 224, 103–116. [Google Scholar] [CrossRef]
- Demaison, L.; Sergiel, J.P.; Moreau, D.; Grynberg, A. Influence of the phospholipid n-6/n-3 polyunsaturated fatty acid ratio on the mitochondrial oxidative metabolism before and after myocardial ischemia. Biochim. Biophys. Acta 1994, 1227, 53–59. [Google Scholar] [CrossRef]
- Abdukeyum, G.G.; Owen, A.J.; McLennan, P.L. Dietary (n-3) long-chain polyunsaturated fatty acids inhibit ischemia and reperfusion arrhythmias and infarction in rat heart not enhanced by ischemic preconditioning. J. Nutr. 2008, 138, 1902–1909. [Google Scholar] [CrossRef] [Green Version]
- Zeghichi-Hamri, S.; de Lorgeril, M.; Salen, P.; Chibane, M.; de Leiris, J.; Boucher, F.; Laporte, F. Protective effect of dietary n-3 polyunsaturated fatty acids on myocardial resistance to ischemia-reperfusion injury in rats. Nutr. Res. 2010, 30, 849–857. [Google Scholar] [CrossRef]
- Pepe, S.; McLennan, P.L. Cardiac membrane fatty acid composition modulates myocardial oxygen consumption and postischemic recovery of contractile function. Circulation 2002, 105, 2303–2308. [Google Scholar] [CrossRef]
- Darwesh, A.M.; Sosnowski, D.K.; Lee, T.Y.; Keshavarz-Bahaghighat, H.; Seubert, J.M. Insights into the cardioprotective properties of n-3 PUFAs against ischemic heart disease via modulation of the innate immune system. Chem. Biol. Interact. 2019, 308, 20–44. [Google Scholar] [CrossRef]
- Ma, D.W.; Seo, J.; Switzer, K.C.; Fan, Y.Y.; McMurray, D.N.; Lupton, J.R.; Chapkin, R.S. n-3 PUFA and membrane microdomains: A new frontier in bioactive lipid research. J. Nutr. Biochem. 2004, 15, 700–706. [Google Scholar] [CrossRef]
- Nuno, D.W.; Coppey, L.J.; Yorek, M.A.; Lamping, K.G. Dietary fats modify vascular fat composition, eNOS localization within lipid rafts and vascular function in obesity. Physiol. Rep. 2018, 6, e13820. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, Q.; Wang, M.; Zhao, S.; Ma, J.; Luo, N.; Li, N.; Li, Y.; Xu, G.; Li, J. Eicosapentaenoic acid modifies lipid composition in caveolae and induces translocation of endothelial nitric oxide synthase. Biochimie 2007, 89, 169–177. [Google Scholar] [CrossRef]
- Yaqoob, P. The nutritional significance of lipid rafts. Annu. Rev. Nutr. 2009, 29, 257–282. [Google Scholar] [CrossRef]
- Chapkin, R.S.; McMurray, D.N.; Davidson, L.A.; Patil, B.S.; Fan, Y.Y.; Lupton, J.R. Bioactive dietary long-chain fatty acids: Emerging mechanisms of action. Br. J. Nutr. 2008, 100, 1152–1157. [Google Scholar] [CrossRef]
- Layne, J.; Majkova, Z.; Smart, E.J.; Toborek, M.; Hennig, B. Caveolae: A regulatory platform for nutritional modulation of inflammatory diseases. J. Nutr. Biochem. 2011, 22, 807–811. [Google Scholar] [CrossRef] [Green Version]
- Shaikh, S.R.; Jolly, C.A.; Chapkin, R.S. n-3 Polyunsaturated fatty acids exert immunomodulatory effects on lymphocytes by targeting plasma membrane molecular organization. Mol. Asp. Med. 2012, 33, 46–54. [Google Scholar] [CrossRef] [Green Version]
- Radosinska, J.; Kurahara, L.H.; Hiraishi, K.; Viczenczova, C.; Egan Benova, T.; Szeiffova Bacova, B.; Dosenko, V.; Navarova, J.; Obsitnik, B.; Imanaga, I.; et al. Modulation of cardiac connexin-43 by omega-3 fatty acid ethyl-ester supplementation demonstrated in spontaneously diabetic rats. Physiol. Res. 2015, 64, 795–806. [Google Scholar] [CrossRef]
- Chung, T.H.; Wang, S.M.; Liang, J.Y.; Yang, S.H.; Wu, J.C. The interaction of estrogen receptor alpha and caveolin-3 regulates connexin43 phosphorylation in metabolic inhibition-treated rat cardiomyocytes. Int. J. Biochem. Cell Biol. 2009, 41, 2323–2333. [Google Scholar] [CrossRef]
- Yang, K.C.; Rutledge, C.A.; Mao, M.; Bakhshi, F.R.; Xie, A.; Liu, H.; Bonini, M.G.; Patel, H.H.; Minshall, R.D.; Dudley, S.C., Jr. Caveolin-1 modulates cardiac gap junction homeostasis and arrhythmogenecity by regulating cSrc tyrosine kinase. Circ. Arrhythm. Electrophysiol. 2014, 7, 701–710. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, E.M.; Pennington, E.R.; Green, W.D.; Beck, M.A.; Brown, D.A.; Shaikh, S.R. Mechanisms by Which Dietary Fatty Acids Regulate Mitochondrial Structure-Function in Health and Disease. Adv. Nutr. 2018, 9, 247–262. [Google Scholar] [CrossRef] [Green Version]
- Shaikh, S.R.; Kinnun, J.J.; Leng, X.; Williams, J.A.; Wassall, S.R. How polyunsaturated fatty acids modify molecular organization in membranes: Insight from NMR studies of model systems. Biochim. Biophys. Acta 2015, 1848, 211–219. [Google Scholar] [CrossRef] [Green Version]
- Rockett, B.D.; Teague, H.; Harris, M.; Melton, M.; Williams, J.; Wassall, S.R.; Shaikh, S.R. Fish oil increases raft size and membrane order of B cells accompanied by differential effects on function. J. Lipid Res. 2012, 53, 674–685. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.A.; Batten, S.E.; Harris, M.; Rockett, B.D.; Shaikh, S.R.; Stillwell, W.; Wassall, S.R. Docosahexaenoic and eicosapentaenoic acids segregate differently between raft and nonraft domains. Biophys. J. 2012, 103, 228–237. [Google Scholar] [CrossRef] [Green Version]
- Lei, S.; Li, H.; Xu, J.; Liu, Y.; Gao, X.; Wang, J.; Ng, K.F.; Lau, W.B.; Ma, X.L.; Rodrigues, B.; et al. Hyperglycemia-induced protein kinase C β2 activation induces diastolic cardiac dysfunction in diabetic rats by impairing caveolin-3 expression and Akt/eNOS signaling. Diabetes 2013, 62, 2318–2328. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Yao, W.; Liu, Z.; Xu, A.; Huang, Y.; Ma, X.L.; Irwin, M.G.; Xia, Z. Hyperglycemia Abrogates Ischemic Postconditioning Cardioprotection by Impairing AdipoR1/Caveolin-3/STAT3 Signaling in Diabetic Rats. Diabetes 2016, 65, 942–955. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Jin, J.; Qiao, S.; Lei, S.; Liao, S.; Ge, Z.D.; Li, H.; Wong, G.T.; Irwin, M.G.; Xia, Z. Inhibition of PKCβ2 overexpression ameliorates myocardial ischaemia/reperfusion injury in diabetic rats via restoring caveolin-3/Akt signaling. Clin. Sci. (Lond.) 2015, 129, 331–344. [Google Scholar] [CrossRef] [Green Version]
- Knowles, C.J.; Cebova, M.; Pinz, I.M. Palmitate diet-induced loss of cardiac caveolin-3: A novel mechanism for lipid-induced contractile dysfunction. PLoS ONE 2013, 8, e61369. [Google Scholar] [CrossRef] [Green Version]
- Knowles, C.J.; Dionne, M.; Cebova, M.; Pinz, I.M. Palmitate-Induced Translocation of Caveolin-3 and Endothelial Nitric Oxide Synthase in Cardiomyocytes. Online J. Biol. Sci. 2011, 11, 27–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, L.; Liu, Q.; Zhang, Q.; Tuo, X.; Fan, D.; Deng, J.; Yang, H. Kiwifruit seed oil prevents obesity by regulating inflammation, thermogenesis, and gut microbiota in high-fat diet-induced obese C57BL/6 mice. Food Chem. Toxicol. 2018, 125, 85–94. [Google Scholar] [CrossRef]
- Marso, S.P.; Miller, T.; Rutherford, B.D.; Gibbons, R.J.; Qureshi, M.; Kalynych, A.; Turco, M.; Schultheiss, H.P.; Mehran, R.; Krucoff, M.W.; et al. Comparison of myocardial reperfusion in patients undergoing percutaneous coronary intervention in ST-segment elevation acute myocardial infarction with versus without diabetes mellitus (from the EMERALD Trial). Am. J. Cardiol. 2007, 100, 206–210. [Google Scholar] [CrossRef]
- Miki, T.; Itoh, T.; Sunaga, D.; Miura, T. Effects of diabetes on myocardial infarct size and cardioprotection by preconditioning and postconditioning. Cardiovasc. Diabetol. 2012, 11. [Google Scholar] [CrossRef] [Green Version]
- Ha, H.; Pak, Y. Modulation of the caveolin-3 and Akt status in caveolae by insulin resistance in H9c2 cardiomyoblasts. Exp. Mol. Med. 2005, 37, 169–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Q.; Gao, F.; Ma, X.L. Insulin says NO to cardiovascular disease. Cardiovasc. Res. 2011, 89, 516–524. [Google Scholar] [CrossRef] [Green Version]
- Schilling, J.M.; Roth, D.M.; Patel, H.H. Caveolins in cardioprotection—Translatability and mechanisms. Br. J. Pharmacol. 2015, 172, 2114–2125. [Google Scholar] [CrossRef] [Green Version]
- See Hoe, L.E.; Schilling, J.M.; Tarbit, E.; Kiessling, C.J.; Busija, A.R.; Niesman, I.R.; Du Toit, E.; Ashton, K.J.; Roth, D.M.; Headrick, J.P.; et al. Sarcolemmal cholesterol and caveolin-3 dependence of cardiac function, ischemic tolerance, and opioidergic cardioprotection. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H895–H903. [Google Scholar] [CrossRef] [Green Version]
- See Hoe, L.E.; May, L.T.; Headrick, J.P.; Peart, J.N. Sarcolemmal dependence of cardiac protection and stress-resistance: Roles in aged or diseased hearts. Br. J. Pharmacol. 2016, 173, 2966–2991. [Google Scholar] [CrossRef] [PubMed]
- Russell, J.; Du Toit, E.F.; Peart, J.N.; Patel, H.H.; Headrick, J.P. Myocyte membrane and microdomain modifications in diabetes: Determinants of ischemic tolerance and cardioprotection. Cardiovasc. Diabetol. 2017, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peart, J.N.; Pepe, S.; Reichelt, M.E.; Beckett, N.; See Hoe, L.; Ozberk, V.; Niesman, I.R.; Patel, H.H.; Headrick, J.P. Dysfunctional survival-signaling and stress-intolerance in aged murine and human myocardium. Exp. Gerontol. 2014, 50, 72–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, J.S.; Griffith, T.A.; Helman, T.; Du Toit, E.F.; Peart, J.N.; Headrick, J.P. Chronic type 2 but not type 1 diabetes impairs myocardial ischaemic tolerance and preconditioning in C57Bl/6 mice. Exp. Physiol. 2019, 104, 1868–1880. [Google Scholar] [CrossRef]
- Russell, J.S.; Budiono, B.P.; Schilling, J.M.; Zemljic-Harpf, A.E.; Patel, H.H.; Peart, J.N.; Headrick, J.P. Abstract 19809: Chronic Type 2 but not Type 1 Diabetes Impairs Myocardial Ischemic Tolerance and Cardioprotection: Effects Countered by Calorie Restriction. In Proceedings of the American Heart Association Scientific Sessions, Anaheim, CA, USA, 11–15 November 2017. [Google Scholar]
- Carotenuto, F.; Minieri, M.; Monego, G.; Fiaccavento, R.; Bertoni, A.; Sinigaglia, F.; Vecchini, A.; Carosella, L.; Di Nardo, P. A diet supplemented with ALA-rich flaxseed prevents cardiomyocyte apoptosis by regulating caveolin-3 expression. Cardiovasc. Res. 2013, 100, 422–431. [Google Scholar] [CrossRef] [Green Version]
- Kukoba, T.V.; Shysh, A.M.; Moibenko, O.O.; Kotsiuruba, A.V.; Kharchenko, O.V. The effects of alpha-linolenic acid on the functioning of the isolated heart during acute myocardial ischemia/reperfusion. Fiziol. Zh. 2006, 52, 12–20. [Google Scholar]
- Farias, J.G.; Carrasco-Pozo, C.; Carrasco Loza, R.; Sepulveda, N.; Alvarez, P.; Quezada, M.; Quinones, J.; Molina, V.; Castillo, R.L. Polyunsaturated fatty acid induces cardioprotection against ischemia-reperfusion through the inhibition of NF-kappaB and induction of Nrf2. Exp. Biol. Med. (Maywood) 2017, 242, 1104–1114. [Google Scholar] [CrossRef] [Green Version]
- Ander, B.P.; Weber, A.R.; Rampersad, P.P.; Gilchrist, J.S.; Pierce, G.N.; Lukas, A. Dietary flaxseed protects against ventricular fibrillation induced by ischemia-reperfusion in normal and hypercholesterolemic Rabbits. J. Nutr. 2004, 134, 3250–3256. [Google Scholar] [CrossRef] [Green Version]
- Creus, A.; Ferreira, M.R.; Oliva, M.E.; Lombardo, Y.B. Mechanisms Involved in the Improvement of Lipotoxicity and Impaired Lipid Metabolism by Dietary alpha-Linolenic Acid Rich Salvia hispanica L (Salba) Seed in the Heart of Dyslipemic Insulin-Resistant Rats. J. Clin. Med. 2016, 5, 18. [Google Scholar] [CrossRef] [Green Version]
- Christopher, C.L.; Mathuram, L.N.; Genitta, G.; Cyrus, I.; Jaya Sundar, S. Omega-3 polyunsaturated fatty acids inhibit the accumulation of PAS-positive material in the myocardium of STZ-diabetic wistar rats. Int. J. Cardiol. 2003, 88, 183–190. [Google Scholar] [CrossRef]
- Anna, Z.; Angela, S.; Barbara, B.; Jana, R.; Tamara, B.; Csilla, V.; Victor, D.; Oleksiy, M.; Narcisa, T. Heart-protective effect of n-3 PUFA demonstrated in a rat model of diabetic cardiomyopathy. Mol. Cell. Biochem. 2014, 389, 219–227. [Google Scholar] [CrossRef]
- Xie, N.; Zhang, W.; Li, J.; Liang, H.; Zhou, H.; Duan, W.; Xu, X.; Yu, S.; Zhang, H.; Yi, D. α-Linolenic acid intake attenuates myocardial ischemia/reperfusion injury through anti-inflammatory and anti-oxidative stress effects in diabetic but not normal rats. Arch. Med. Res. 2011, 42, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Seti, H.; Leikin-Frenkel, A.; Werner, H. Effects of omega-3 and omega-6 fatty acids on IGF-I receptor signalling in colorectal cancer cells. Arch. Physiol. Biochem. 2009, 115, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Yan, P.; Zhang, S.; Huang, H.; Huang, F.; Sun, T.; Deng, Q.; Huang, Q.; Chen, S.; Ye, K.; et al. Long-Term Dietary Alpha-Linolenic Acid Supplement Alleviates Cognitive Impairment Correlate with Activating Hippocampal CREB Signaling in Natural Aging Rats. Mol. Neurobiol. 2016, 53, 4772–4786. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Kim, M.H.; Ahn, J.H.; Lee, S.J.; Lee, J.H.; Eum, W.S.; Choi, S.Y.; Kwon, H.Y. The Stimulatory Effect of Essential Fatty Acids on Glucose Uptake Involves Both Akt and AMPK Activation in C2C12 Skeletal Muscle Cells. Korean J. Physiol. Pharmacol. 2014, 18, 255–261. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Deng, Q.; Tang, Y.; Xiao, L.; Liu, L.; Yao, P.; Tang, H.; Dong, X. Flaxseed Oil Attenuates Hepatic Steatosis and Insulin Resistance in Mice by Rescuing the Adaption to ER Stress. J. Agric. Food Chem. 2018, 66, 10729–10740. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, R.; Han, S.F.; Bu, L.; Wang, S.W.; Ma, H.; Jia, G.L. Alpha-linolenic acid attenuates high glucose-induced apoptosis in cultured human umbilical vein endothelial cells via PI3K/Akt/eNOS pathway. Nutrition 2007, 23, 762–770. [Google Scholar] [CrossRef]
- Zhang, W.; Li, R.; Li, J.; Wang, W.; Tie, R.; Tian, F.; Liang, X.; Xing, W.; He, Y.; Yu, L.; et al. Alpha-linolenic acid exerts an endothelial protective effect against high glucose injury via PI3K/Akt pathway. PLoS ONE 2013, 8, e68489. [Google Scholar] [CrossRef]
- Carotenuto, F.; Costa, A.; Albertini, M.C.; Rocchi, M.B.; Rudov, A.; Coletti, D.; Minieri, M.; Di Nardo, P.; Teodori, L. Dietary Flaxseed Mitigates Impaired Skeletal Muscle Regeneration: In Vivo, in Vitro and in Silico Studies. Int. J. Med. Sci. 2016, 13, 206–219. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Lim, E.J.; Toborek, M.; Hennig, B. The role of fatty acids and caveolin-1 in TNF-α-induced endothelial cell activation. Metabolism 2008, 57, 1328–1339. [Google Scholar] [CrossRef] [Green Version]
- Gortan Cappellari, G.; Losurdo, P.; Mazzucco, S.; Panizon, E.; Jevnicar, M.; Macaluso, L.; Fabris, B.; Barazzoni, R.; Biolo, G.; Carretta, R.; et al. Treatment with n-3 polyunsaturated fatty acids reverses endothelial dysfunction and oxidative stress in experimental menopause. J. Nutr. Biochem. 2013, 24, 371–379. [Google Scholar] [CrossRef]
- Fricovsky, E.S.; Suarez, J.; Ihm, S.H.; Scott, B.T.; Suarez-Ramirez, J.A.; Banerjee, I.; Torres-Gonzalez, M.; Wang, H.; Ellrott, I.; Maya-Ramos, L.; et al. Excess protein O-GlcNAcylation and the progression of diabetic cardiomyopathy. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 303, R689–R699. [Google Scholar] [CrossRef]
- Pan, M.; Han, Y.; Basu, A.; Dai, A.; Si, R.; Willson, C.; Balistrieri, A.; Scott, B.T.; Makino, A. Overexpression of hexokinase 2 reduces mitochondrial calcium overload in coronary endothelial cells of type 2 diabetic mice. Am. J. Physiol. Cell Physiol. 2018, 314, C732–C740. [Google Scholar] [CrossRef]
- Skovso, S. Modeling type 2 diabetes in rats using high fat diet and streptozotocin. J. Diabetes Investig. 2014, 5, 349–358. [Google Scholar] [CrossRef]
- Parikh, J.; Zemljic-Harpf, A.; Fu, J.; Giamouridis, D.; Hsieh, T.C.; Kassan, A.; Murthy, K.S.; Bhargava, V.; Patel, H.H.; Rajasekaran, M.R. Altered Penile Caveolin Expression in Diabetes: Potential Role in Erectile Dysfunction. J. Sex. Med. 2017, 14, 1177–1186. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Ma, Z.; Hu, W.; Wang, D.; Jiang, S.; Fan, C.; Di, S.; Liu, D.; Sun, Y.; Yi, W. Caveolin-1/-3: Therapeutic targets for myocardial ischemia/reperfusion injury. Basic Res. Cardiol. 2016, 111, 45. [Google Scholar] [CrossRef]
- Kaakinen, M.; Reichelt, M.E.; Ma, Z.; Ferguson, C.; Martel, N.; Porrello, E.R.; Hudson, J.E.; Thomas, W.G.; Parton, R.G.; Headrick, J.P. Cavin-1 deficiency modifies myocardial and coronary function, stretch responses and ischaemic tolerance: Roles of NOS over-activity. Basic Res. Cardiol. 2017, 112, 24. [Google Scholar] [CrossRef]
- Naito, D.; Ogata, T.; Hamaoka, T.; Nakanishi, N.; Miyagawa, K.; Maruyama, N.; Kasahara, T.; Taniguchi, T.; Nishi, M.; Matoba, S.; et al. The coiled-coil domain of MURC/cavin-4 is involved in membrane trafficking of caveolin-3 in cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H2127–H2136. [Google Scholar] [CrossRef]
- Nishi, M.; Ogata, T.; Cannistraci, C.V.; Ciucci, S.; Nakanishi, N.; Higuchi, Y.; Sakamoto, A.; Tsuji, Y.; Mizushima, K.; Matoba, S. Systems Network Genomic Analysis Reveals Cardioprotective Effect of MURC/Cavin-4 Deletion Against Ischemia/Reperfusion Injury. J. Am. Heart Assoc. 2019, 8, e012047. [Google Scholar] [CrossRef]
- Balogun, K.A.; Albert, C.J.; Ford, D.A.; Brown, R.J.; Cheema, S.K. Dietary omega-3 polyunsaturated fatty acids alter the fatty acid composition of hepatic and plasma bioactive lipids in C57BL/6 mice: A lipidomic approach. PLoS ONE 2013, 8, e82399. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Muniyappa, R.; Yan, X.; Chen, H.; Yue, L.Q.; Hong, E.G.; Kim, J.K.; Quon, M.J. Comparison between surrogate indexes of insulin sensitivity and resistance and hyperinsulinemic euglycemic clamp estimates in mice. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E261–E270. [Google Scholar] [CrossRef] [Green Version]
- Cacho, J.; Sevillano, J.; de Castro, J.; Herrera, E.; Ramos, M.P. Validation of simple indexes to assess insulin sensitivity during pregnancy in Wistar and Sprague-Dawley rats. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E1269–E1276. [Google Scholar] [CrossRef]
- Reichelt, M.E.; Willems, L.; Hack, B.A.; Peart, J.N.; Headrick, J.P. Cardiac and coronary function in the Langendorff-perfused mouse heart model. Exp. Physiol. 2009, 94, 54–70. [Google Scholar] [CrossRef]
- Tsang, A.; Hausenloy, D.J.; Mocanu, M.M.; Carr, R.D.; Yellon, D.M. Preconditioning the diabetic heart: The importance of Akt phosphorylation. Diabetes 2005, 54, 2360–2364. [Google Scholar] [CrossRef] [Green Version]
- Peart, J.; Headrick, J.P. Adenosine-mediated early preconditioning in mouse: Protective signaling and concentration dependent effects. Cardiovasc. Res. 2003, 58, 589–601. [Google Scholar] [CrossRef] [Green Version]
- Rana, A.; Sharma, S. Mechanism of sphingosine-1-phosphate induced cardioprotection against I/R injury in diabetic rat heart: Possible involvement of glycogen synthase kinase 3 and mitochondrial permeability transition pore. Clin. Exp. Pharmacol. Physiol. 2016, 43, 166–173. [Google Scholar] [CrossRef]
- Abedinpour, P.; Jergil, B. Isolation of a caveolae-enriched fraction from rat lung by affinity partitioning and sucrose gradient centrifugation. Anal. Biochem. 2002, 313, 1–8. [Google Scholar] [CrossRef]
- Lisanti, M.P.; Scherer, J.P.; Vidugiriene, J.; Tang, Z.; Hermanowski-Vosatka, A.; Tu, Y.H.; Cook, R.F.; Sargiacomo, M. Characterization of caveolin-rich membrane domains isolated from an endothelial-rich source: Implications for human disease. J. Cell Biol. 1994, 126, 111–126. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Ludbrook, J. On making multiple comparisons in clinical and experimental pharmacology and physiology. Clin. Exp. Pharmacol. Physiol. 1991, 18, 379–392. [Google Scholar] [CrossRef]
- Kusakabe, T.; Tanioka, H.; Ebihara, K.; Hirata, M.; Miyamoto, L.; Miyanaga, F.; Hige, H.; Aotani, D.; Fujisawa, T.; Masuzaki, H.; et al. Beneficial effects of leptin on glycaemic and lipid control in a mouse model of type 2 diabetes with increased adiposity induced by streptozotocin and a high-fat diet. Diabetologia 2009, 52, 675–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, Y.E.; Basu, A.; Dai, A.; Heldak, M.; Makino, A. Coronary endothelial dysfunction and mitochondrial reactive oxygen species in type 2 diabetic mice. Am. J. Physiol. Cell Physiol. 2013, 305, C1033–C1040. [Google Scholar] [CrossRef] [Green Version]
- Kolocassides, K.G.; Seymour, A.M.; Galinanes, M.; Hearse, D.J. Paradoxical effect of ischemic preconditioning on ischemic contracture? NMR studies of energy metabolism and intracellular pH in the rat heart. J. Mol. Cell Cardiol. 1996, 28, 1045–1057. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Cui, L.; Zhang, Z.; Zhao, Q.; Li, S. alpha-Linolenic acid attenuates doxorubicin-induced cardiotoxicity in rats through suppression of oxidative stress and apoptosis. Acta Biochim. Biophys. Sin. (Shanghai) 2013, 45, 817–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Black, S.C.; Katz, S.; McNeill, J.H. Cardiac performance and plasma lipids of omega-3 fatty acid-treated streptozocin-induced diabetic rats. Diabetes 1989, 38, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, D.; Helies-Toussaint, C.; Moreau, D.; Raederstorff, D.; Grynberg, A. Dietary n-3 PUFAs affect the blood pressure rise and cardiac impairments in a hyperinsulinemia rat model in vivo. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H1294–H1302. [Google Scholar] [CrossRef] [Green Version]
- Tuncay, E.; Seymen, A.A.; Tanriverdi, E.; Yaras, N.; Tandogan, B.; Ulusu, N.N.; Turan, B. Gender related differential effects of Omega-3E treatment on diabetes-induced left ventricular dysfunction. Mol. Cell. Biochem. 2007, 304, 255–263. [Google Scholar] [CrossRef]
- Patel, T.P.; Rawal, K.; Bagchi, A.K.; Akolkar, G.; Bernardes, N.; Dias, D.D.S.; Gupta, S.; Singal, P.K. Insulin resistance: An additional risk factor in the pathogenesis of cardiovascular disease in type 2 diabetes. Heart Fail. Rev. 2016, 21, 11–23. [Google Scholar] [CrossRef]
- Hartweg, J.; Perera, R.; Montori, V.; Dinneen, S.; Neil, H.A.; Farmer, A. Omega-3 polyunsaturated fatty acids (PUFA) for type 2 diabetes mellitus. Cochrane Database Syst. Rev. 2008, 1, CD003205. [Google Scholar] [CrossRef]
- Vessby, B.; Karlstrom, B.; Boberg, M.; Lithell, H.; Berne, C. Polyunsaturated fatty acids may impair blood glucose control in type 2 diabetic patients. Diabet. Med. 1992, 9, 126–133. [Google Scholar] [CrossRef]
- Field, C.J.; Ryan, E.A.; Thomson, A.B.; Clandinin, M.T. Diet fat composition alters membrane phospholipid composition, insulin binding, and glucose metabolism in adipocytes from control and diabetic animals. J. Biol. Chem. 1990, 265, 11143–11150. [Google Scholar]
- Coelho, O.G.L.; da Silva, B.P.; Rocha, D.; Lopes, L.L.; Alfenas, R.C.G. Polyunsaturated fatty acids and type 2 diabetes: Impact on the glycemic control mechanism. Crit. Rev. Food Sci. Nutr. 2017, 57, 3614–3619. [Google Scholar] [CrossRef] [PubMed]
- Imamura, F.; Micha, R.; Wu, J.H.; de Oliveira Otto, M.C.; Otite, F.O.; Abioye, A.I.; Mozaffarian, D. Effects of Saturated Fat, Polyunsaturated Fat, Monounsaturated Fat, and Carbohydrate on Glucose-Insulin Homeostasis: A Systematic Review and Meta-analysis of Randomised Controlled Feeding Trials. PLoS Med. 2016, 13, e1002087. [Google Scholar] [CrossRef] [Green Version]
- Heskey, C.E.; Jaceldo-Siegl, K.; Sabate, J.; Fraser, G.; Rajaram, S. Adipose tissue alpha-linolenic acid is inversely associated with insulin resistance in adults. Am. J. Clin. Nutr. 2016, 103, 1105–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sala-Vila, A.; Guasch-Ferre, M.; Hu, F.B.; Sanchez-Tainta, A.; Bullo, M.; Serra-Mir, M.; Lopez-Sabater, C.; Sorli, J.V.; Aros, F.; Fiol, M.; et al. Dietary alpha-Linolenic Acid, Marine omega-3 Fatty Acids, and Mortality in a Population With High Fish Consumption: Findings From the PREvencion con DIeta MEDiterranea (PREDIMED) Study. J. Am. Heart. Assoc. 2016, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamat, S.G.; Roy, R. Evaluation of the effect of n-3 PUFA-rich dietary fish oils on lipid profile and membrane fluidity in alloxan-induced diabetic mice (Mus musculus). Mol. Cell. Biochem. 2016, 416, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Black, S.C.; Katz, S.; McNeill, J.H. Influence of omega-3 fatty acid treatment on cardiac phospholipid composition and coronary flow of streptozocin-diabetic rats. Metabolism 1993, 42, 320–326. [Google Scholar] [CrossRef]
- Coppey, L.; Davidson, E.; Shevalye, H.; Obrosov, A.; Yorek, M. Effect of Early and Late Interventions with Dietary Oils on Vascular and Neural Complications in a Type 2 Diabetic Rat Model. J. Diabetes Res. 2019, 2019, 5020465. [Google Scholar] [CrossRef] [Green Version]
- Shevalye, H.; Yorek, M.S.; Coppey, L.J.; Holmes, A.; Harper, M.M.; Kardon, R.H.; Yorek, M.A. Effect of enriching the diet with menhaden oil or daily treatment with resolvin D1 on neuropathy in a mouse model of type 2 diabetes. J. Neurophysiol. 2015, 114, 199–208. [Google Scholar] [CrossRef] [Green Version]
- Mustad, V.A.; Demichele, S.; Huang, Y.S.; Mika, A.; Lubbers, N.; Berthiaume, N.; Polakowski, J.; Zinker, B. Differential effects of n-3 polyunsaturated fatty acids on metabolic control and vascular reactivity in the type 2 diabetic ob/ob mouse. Metabolism 2006, 55, 1365–1374. [Google Scholar] [CrossRef]
- Coppey, L.J.; Holmes, A.; Davidson, E.P.; Yorek, M.A. Partial replacement with menhaden oil improves peripheral neuropathy in high-fat-fed low-dose streptozotocin type 2 diabetic rat. J. Nutr. Metab. 2012, 2012, 950517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domenichiello, A.F.; Kitson, A.P.; Metherel, A.H.; Chen, C.T.; Hopperton, K.E.; Stavro, P.M.; Bazinet, R.P. Whole-Body Docosahexaenoic Acid Synthesis-Secretion Rates in Rats Are Constant across a Large Range of Dietary alpha-Linolenic Acid Intakes. J. Nutr. 2017, 147, 37–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goo, S.; Han, J.C.; Nisbet, L.A.; Legrice, I.J.; Taberner, A.J.; Loiselle, D.S. Dietary supplementation with either saturated or unsaturated fatty acids does not affect the mechanoenergetics of the isolated rat heart. Physiol. Rep. 2014, 2, e00272. [Google Scholar] [CrossRef] [PubMed]
- Goo, S.; Han, J.C.; Nisbet, L.A.; LeGrice, I.J.; Taberner, A.J.; Loiselle, D.S. Dietary pre-exposure of rats to fish oil does not enhance myocardial efficiency of isolated working hearts or their left ventricular trabeculae. J. Physiol. 2014, 592, 1795–1808. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, K.M.; Khairallah, R.J.; Sparagna, G.C.; Xu, W.; Hecker, P.A.; Robillard-Frayne, I.; Des Rosiers, C.; Kristian, T.; Murphy, R.C.; Fiskum, G.; et al. Dietary omega-3 fatty acids alter cardiac mitochondrial phospholipid composition and delay Ca2+-induced permeability transition. J. Mol. Cell Cardiol. 2009, 47, 819–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pepe, S.; McLennan, P.L. (n-3) Long chain PUFA dose-dependently increase oxygen utilization efficiency and inhibit arrhythmias after saturated fat feeding in rats. J. Nutr. 2007, 137, 2377–2383. [Google Scholar] [CrossRef] [Green Version]
- Force, T.; Malis, C.D.; Guerrero, J.L.; Varadarajan, G.S.; Bonventre, J.V.; Weber, P.C.; Leaf, A. n-3 fatty acids increase postischemic blood flow but do not reduce myocardial necrosis. Am. J. Physiol. 1989, 257, H1204–H1210. [Google Scholar] [CrossRef]
- Zirpoli, H.; Abdillahi, M.; Quadri, N.; Ananthakrishnan, R.; Wang, L.; Rosario, R.; Zhu, Z.; Deckelbaum, R.J.; Ramasamy, R. Acute administration of n-3 rich triglyceride emulsions provides cardioprotection in murine models after ischemia-reperfusion. PLoS ONE 2015, 10, e0116274. [Google Scholar] [CrossRef] [Green Version]
- Endo, J.; Arita, M. Cardioprotective mechanism of omega-3 polyunsaturated fatty acids. J. Cardiol. 2016, 67, 22–27. [Google Scholar] [CrossRef] [Green Version]
- Flachs, P.; Rossmeisl, M.; Kopecky, J. The effect of n-3 fatty acids on glucose homeostasis and insulin sensitivity. Physiol. Res. 2014, 63 (Suppl. 1), S93–S118. [Google Scholar] [CrossRef]
- McLennan, P.L. Cardiac physiology and clinical efficacy of dietary fish oil clarified through cellular mechanisms of omega-3 polyunsaturated fatty acids. Eur. J. Appl. Physiol. 2014, 114, 1333–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horikawa, Y.T.; Patel, H.H.; Tsutsumi, Y.M.; Jennings, M.M.; Kidd, M.W.; Hagiwara, Y.; Ishikawa, Y.; Insel, P.A.; Roth, D.M. Caveolin-3 expression and caveolae are required for isoflurane-induced cardiac protection from hypoxia and ischemia/reperfusion injury. J. Mol. Cell. Cardiol. 2008, 44, 123–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busija, A.R.; Patel, H.H.; Insel, P.A. Caveolins and cavins in the trafficking, maturation, and degradation of caveolae: Implications for cell physiology. Am. J. Physiol. Cell Physiol. 2017, 312, C459–C477. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Hu, X.; Lee, S.H.; Chen, F.; Jiang, K.; Tu, Z.; Liu, Z.; Du, J.; Wang, L.; Yin, C.; et al. Diabetes Exacerbates Myocardial Ischemia/Reperfusion Injury by Down-Regulation of MicroRNA and Up-Regulation of O-GlcNAcylation. JACC Basic Transl. Sci. 2018, 3, 350–362. [Google Scholar] [CrossRef]
- Wang, J.; Schilling, J.M.; Niesman, I.R.; Headrick, J.P.; Finley, J.C.; Kwan, E.; Patel, P.M.; Head, B.P.; Roth, D.M.; Yue, Y.; et al. Cardioprotective trafficking of caveolin to mitochondria is Gi-protein dependent. Anesthesiology 2014, 121, 538–548. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Zhang, Q.; Wang, M.; Liu, F.; Zhao, S.; Ma, J.; Luo, N.; Li, N.; Li, Y.; Xu, G.; et al. Docosahexaenoic acid affects endothelial nitric oxide synthase in caveolae. Arch. Biochem. Biophys. 2007, 466, 250–259. [Google Scholar] [CrossRef]
- Stary, C.M.; Tsutsumi, Y.M.; Patel, P.M.; Head, B.P.; Patel, H.H.; Roth, D.M. Caveolins: Targeting pro-survival signaling in the heart and brain. Front Physiol. 2012, 3, 393. [Google Scholar] [CrossRef] [Green Version]
- Moreira, J.D.; Knorr, L.; Thomazi, A.P.; Simao, F.; Battu, C.; Oses, J.P.; Gottfried, C.; Wofchuk, S.; Salbego, C.; Souza, D.O.; et al. Dietary omega-3 fatty acids attenuate cellular damage after a hippocampal ischemic insult in adult rats. J. Nutr. Biochem. 2010, 21, 351–356. [Google Scholar] [CrossRef]
- Lim, K.; Han, C.; Dai, Y.; Shen, M.; Wu, T. Omega-3 polyunsaturated fatty acids inhibit hepatocellular carcinoma cell growth through blocking beta-catenin and cyclooxygenase-2. Mol. Cancer Ther. 2009, 8, 3046–3055. [Google Scholar] [CrossRef] [Green Version]
- Boengler, K.; Lochnit, G.; Schulz, R. Mitochondria “THE” target of myocardial conditioning. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1215–H1231. [Google Scholar] [CrossRef] [Green Version]
- Panasiuk, O.; Shysh, A.; Bondarenko, A.; Moibenko, O. Omega-3 polyunsaturated fatty acid-enriched diet differentially protects two subpopulations of myocardial mitochondria against Ca(2+)-induced injury. Exp. Clin. Cardiol. 2013, 18, e60–e64. [Google Scholar] [PubMed]
- Sullivan, E.M.; Pennington, E.R.; Sparagna, G.C.; Torres, M.J.; Neufer, P.D.; Harris, M.; Washington, J.; Anderson, E.J.; Zeczycki, T.N.; Brown, D.A.; et al. Docosahexaenoic acid lowers cardiac mitochondrial enzyme activity by replacing linoleic acid in the phospholipidome. J. Biol. Chem. 2018, 293, 466–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ovide-Bordeaux, S.; Grynberg, A. Docosahexaenoic acid affects insulin deficiency- and insulin resistance-induced alterations in cardiac mitochondria. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 286, R519–R527. [Google Scholar] [CrossRef] [Green Version]
- Shoelson, S.E.; Herrero, L.; Naaz, A. Obesity, inflammation, and insulin resistance. Gastroenterology 2007, 132, 2169–2180. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Inflammation and endoplasmic reticulum stress in obesity and diabetes. Int. J. Obes. (Lond.) 2008, 32 (Suppl. 7), S52–S54. [Google Scholar] [CrossRef] [Green Version]
- Demirtas, L.; Guclu, A.; Erdur, F.M.; Akbas, E.M.; Ozcicek, A.; Onk, D.; Turkmen, K. Apoptosis, autophagy & endoplasmic reticulum stress in diabetes mellitus. Indian J. Med. Res. 2016, 144, 515–524. [Google Scholar] [CrossRef]
- Fan, R.; Kim, J.; You, M.; Giraud, D.; Toney, A.M.; Shin, S.H.; Kim, S.Y.; Borkowski, K.; Newman, J.W.; Chung, S. alpha-Linolenic acid-enriched butter attenuated high fat diet-induced insulin resistance and inflammation by promoting bioconversion of n-3 PUFA and subsequent oxylipin formation. J. Nutr. Biochem. 2020, 76, 108285. [Google Scholar] [CrossRef]
- Liu, Y.; Paterson, M.; Baumgardt, S.L.; Irwin, M.G.; Xia, Z.; Bosnjak, Z.J.; Ge, Z.D. Vascular endothelial growth factor regulation of endothelial nitric oxide synthase phosphorylation is involved in isoflurane cardiac preconditioning. Cardiovasc. Res. 2019, 115, 168–178. [Google Scholar] [CrossRef]
- Hong, X.Y.; Hong, X.; Gu, W.W.; Lin, J.; Yin, W.T. Cardioprotection and improvement in endothelial-dependent vasodilation during late-phase of whole body hypoxic preconditioning in spontaneously hypertensive rats via VEGF and endothelin-1. Eur. J. Pharmacol. 2019, 842, 79–88. [Google Scholar] [CrossRef]
- Cohen, M.V.; Downey, J.M. Signalling pathways and mechanisms of protection in pre- and postconditioning: Historical perspective and lessons for the future. Br. J. Pharmacol. 2015, 172, 1913–1932. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.H.; Han, Q.F.; Gao, L.; Sun, Y.; Tang, Z.W.; Wang, M.; Wang, W.; Yao, H.C. HMGB1 Protects the Heart Against Ischemia-Reperfusion Injury via PI3K/AkT Pathway-Mediated Upregulation of VEGF Expression. Front. Physiol. 2019, 10, 1595. [Google Scholar] [CrossRef] [PubMed]
- Kones, R.; Howell, S.; Rumana, U. n-3 Polyunsaturated Fatty Acids and Cardiovascular Disease: Principles, Practices, Pitfalls, and Promises-A Contemporary Review. Med. Princ. Pract. 2017, 26, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.; Konkel, A.; Mehling, H.; Blossey, K.; Gapelyuk, A.; Wessel, N.; von Schacky, C.; Dechend, R.; Muller, D.N.; Rothe, M.; et al. Dietary omega-3 fatty acids modulate the eicosanoid profile in man primarily via the CYP-epoxygenase pathway. J. Lipid Res. 2014, 55, 1150–1164. [Google Scholar] [CrossRef] [Green Version]
- Wainwright, C.L.; Michel, L. Endocannabinoid system as a potential mechanism for n-3 long-chain polyunsaturated fatty acid mediated cardiovascular protection. Proc. Nutr. Soc. 2013, 72, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Di Nunzio, M.; Danesi, F.; Bordoni, A. n-3 PUFA as regulators of cardiac gene transcription: A new link between PPAR activation and fatty acid composition. Lipids 2009, 44, 1073–1079. [Google Scholar] [CrossRef] [PubMed]
- Sakai, C.; Ishida, M.; Ohba, H.; Yamashita, H.; Uchida, H.; Yoshizumi, M.; Ishida, T. Fish oil omega-3 polyunsaturated fatty acids attenuate oxidative stress-induced DNA damage in vascular endothelial cells. PLoS ONE 2017, 12, e0187934. [Google Scholar] [CrossRef] [Green Version]
- Schulz, R.; Gorge, P.M.; Gorbe, A.; Ferdinandy, P.; Lampe, P.D.; Leybaert, L. Connexin 43 is an emerging therapeutic target in ischemia/reperfusion injury, cardioprotection and neuroprotection. Pharmacol. Ther. 2015, 153, 90–106. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Petzke, M.M.; Iyer, R.; Wu, H.; Schwartz, I. Pattern of proinflammatory cytokine induction in RAW264.7 mouse macrophages is identical for virulent and attenuated Borrelia burgdorferi. J. Immunol. 2008, 180, 8306–8315. [Google Scholar] [CrossRef]
- Im, S.S.; Yousef, L.; Blaschitz, C.; Liu, J.Z.; Edwards, R.A.; Young, S.G.; Raffatellu, M.; Osborne, T.F. Linking lipid metabolism to the innate immune response in macrophages through sterol regulatory element binding protein-1a. Cell Metab. 2011, 13, 540–549. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, H.; Omelchenko, I.; Shi, X.; Nuttall, A.L. The influence of NF-kappaB signal-transduction pathways on the murine inner ear by acoustic overstimulation. J. Neurosci. Res. 2009, 87, 1832–1840. [Google Scholar] [CrossRef] [Green Version]
- Yamakawa, I.; Kojima, H.; Terashima, T.; Katagi, M.; Oi, J.; Urabe, H.; Sanada, M.; Kawai, H.; Chan, L.; Yasuda, H.; et al. Inactivation of TNF-alpha ameliorates diabetic neuropathy in mice. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E844–E852. [Google Scholar] [CrossRef] [PubMed]
- McClellan, S.; Jiang, X.; Barrett, R.; Hazlett, L.D. High-mobility group box 1: A novel target for treatment of Pseudomonas aeruginosa keratitis. J. Immunol. 2015, 194, 1776–1787. [Google Scholar] [CrossRef] [PubMed]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Russell, J.S.; Griffith, T.A.; Naghipour, S.; Vider, J.; Du Toit, E.F.; Patel, H.H.; Peart, J.N.; Headrick, J.P. Dietary α-Linolenic Acid Counters Cardioprotective Dysfunction in Diabetic Mice: Unconventional PUFA Protection. Nutrients 2020, 12, 2679. https://doi.org/10.3390/nu12092679
Russell JS, Griffith TA, Naghipour S, Vider J, Du Toit EF, Patel HH, Peart JN, Headrick JP. Dietary α-Linolenic Acid Counters Cardioprotective Dysfunction in Diabetic Mice: Unconventional PUFA Protection. Nutrients. 2020; 12(9):2679. https://doi.org/10.3390/nu12092679
Chicago/Turabian StyleRussell, Jake S., Tia A. Griffith, Saba Naghipour, Jelena Vider, Eugene F. Du Toit, Hemal H. Patel, Jason N. Peart, and John P. Headrick. 2020. "Dietary α-Linolenic Acid Counters Cardioprotective Dysfunction in Diabetic Mice: Unconventional PUFA Protection" Nutrients 12, no. 9: 2679. https://doi.org/10.3390/nu12092679