The Role of Dietary Antioxidants in the Pathogenesis of Neurodegenerative Diseases and Their Impact on Cerebral Oxidoreductive Balance

 ,
,
Abstract
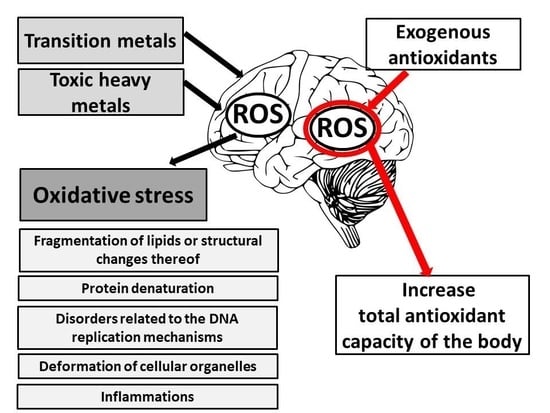
:
1. Introduction
2. The pathogenesis of Neurodegenerative Diseases
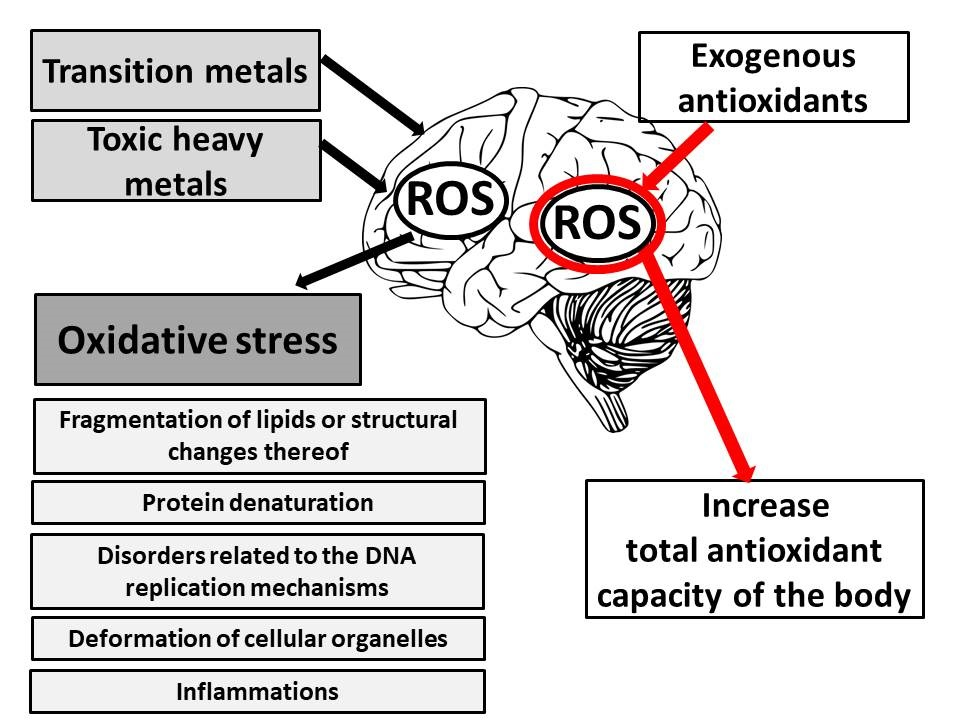
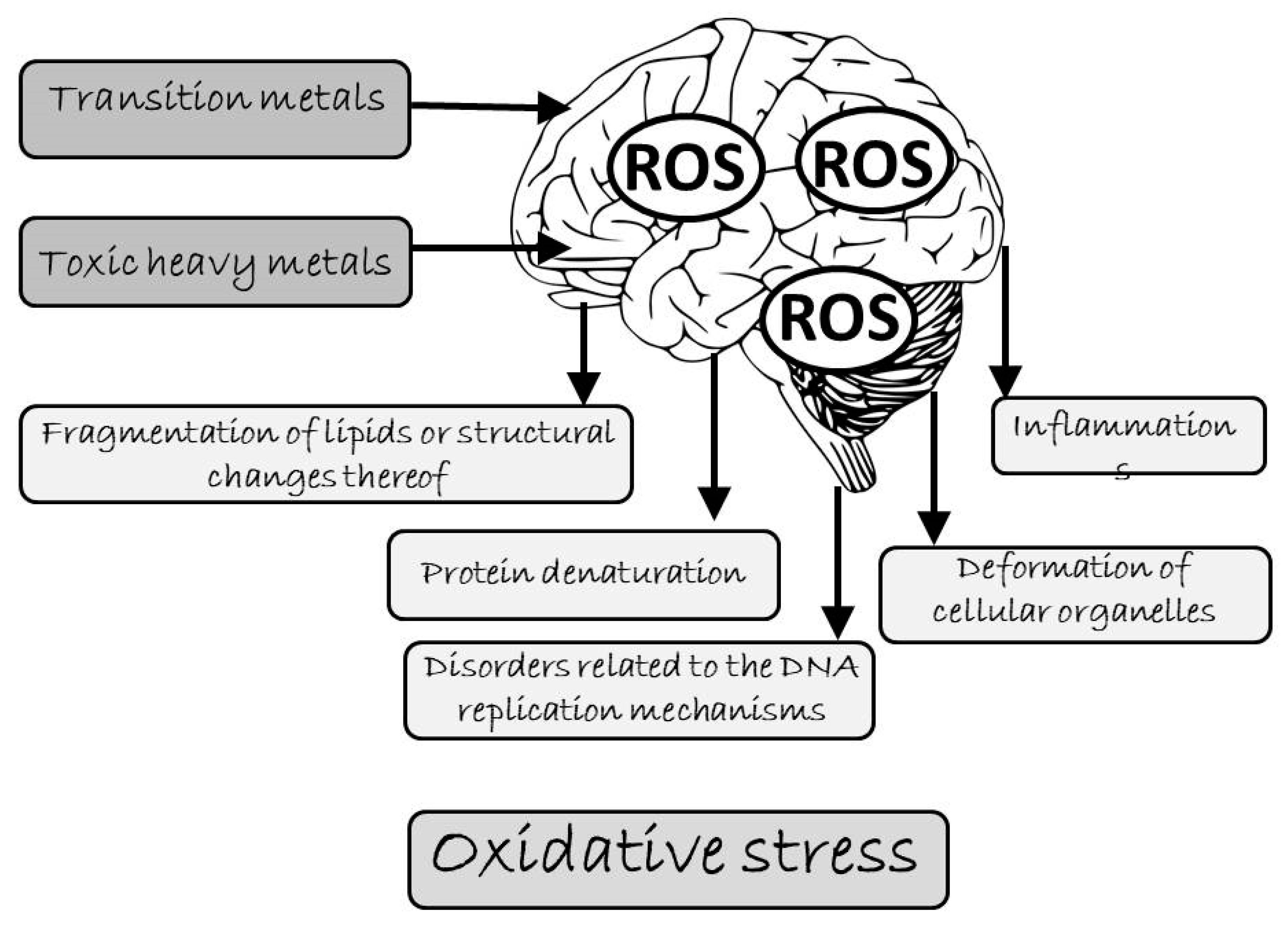
3. Oxidative Stress as the Primary Cause of Brain Damage
4. Ion Metals Stimulating Cellular Oxidation
4.1. Transition Metals
 Cu2+ + e– and Fe2+
Cu2+ + e– and Fe2+  Fe3+ + e–. Their chemical character allows some of the compounds to take part in a range of physiological redox reactions [54]. They are involved in the formation of HO• from H2O2 in the course of Fenton’s reaction (they can catalyze Haer-Weiss reaction) and initiate non-specific lipid peroxidation [10]. The mechanisms of forming free radicals with the participation of transition metal ions are illustrated by Fenton’s and Haber-Weiss reactions [55]. In Fenton’s reaction, hydrogen peroxide disintegrates in the presence of transition metal (Me) ions (Fe2+, Cu2+) creating a hydroxyl radical:
Fe3+ + e–. Their chemical character allows some of the compounds to take part in a range of physiological redox reactions [54]. They are involved in the formation of HO• from H2O2 in the course of Fenton’s reaction (they can catalyze Haer-Weiss reaction) and initiate non-specific lipid peroxidation [10]. The mechanisms of forming free radicals with the participation of transition metal ions are illustrated by Fenton’s and Haber-Weiss reactions [55]. In Fenton’s reaction, hydrogen peroxide disintegrates in the presence of transition metal (Me) ions (Fe2+, Cu2+) creating a hydroxyl radical:4.2. Toxic Heavy Metals
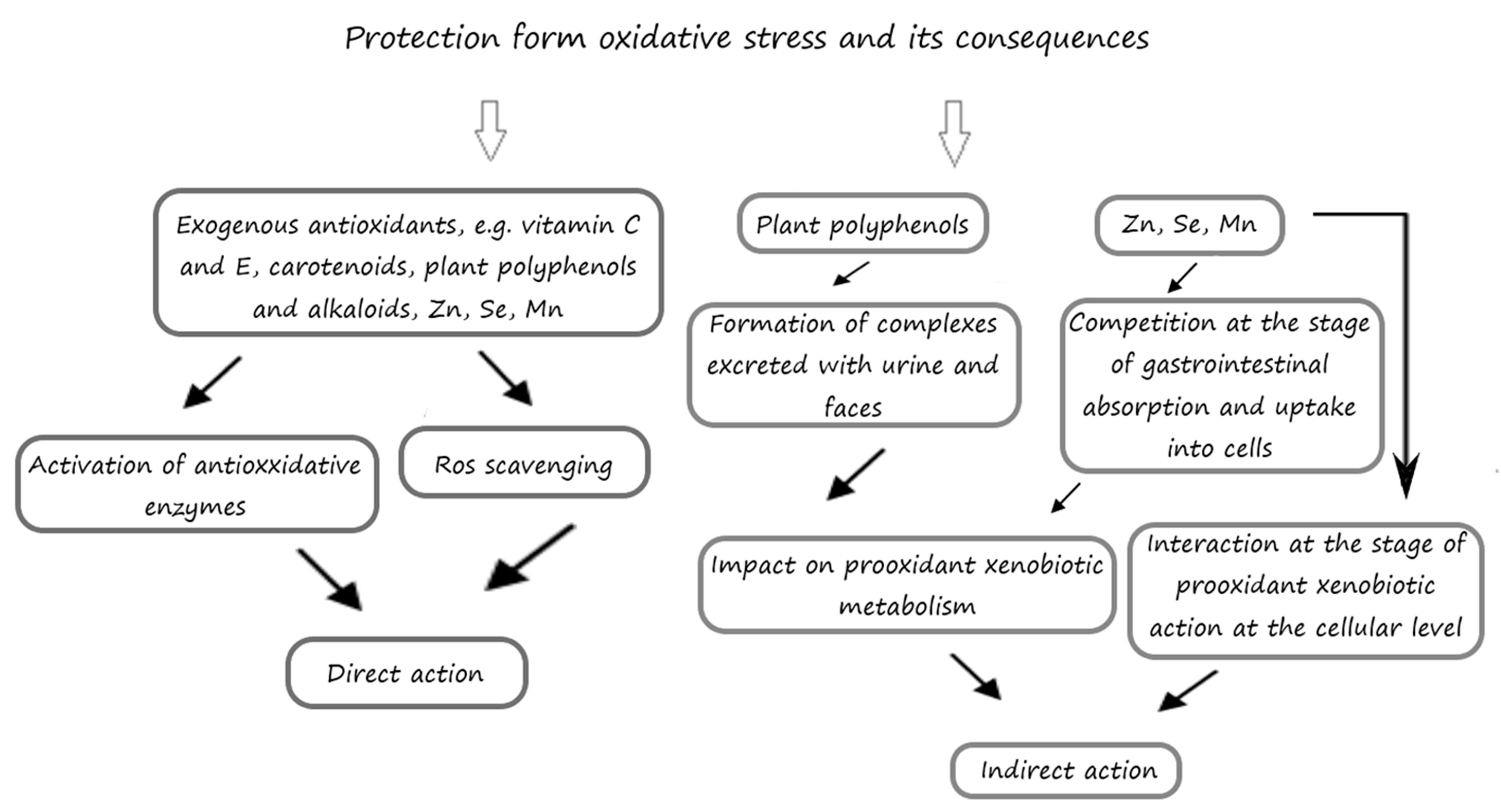
5. The Inhibitory Effects of Exogenous Antioxidants on the Processes of Oxidation
5.1. The Influence of Exogenous Antioxidants on the Cerebral Antioxidative Status in Laboratory Animals
5.1.1. Phenolic Compounds
5.1.2. Alkaloids
5.1.3. Vitamins and Provitamins
6. Antioxidative Therapies in Neurodegenerative Diseases—Clinical Studies
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Łabuzek, K.; Skrudlik, E.; Gabryel, B.; Okopień, B. Anti-inflammatory microglial cell function in the light of the latest scientific research. Ann. Acad. Med. Siles. 2015, 69, 99–110. [Google Scholar] [CrossRef]
- Brown, R.C.; Lockwood, A.H.; Sonawane, B.R. Neurodegenerative diseases, an overview of environmental risk factors. Environ. Health Perspect. 2005, 113, 1250–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kannappan, R.; Gupta, S.C.; Kim, J.H.; Reuter, S.; Aggarwal, B.B. Neuroprotection by spice-derived nutraceuticals, you are what you eat! Mol. Neurobiol. 2011, 44, 142–159. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Majumdar, D.; Gao, Y.; Brewer, B.M.; Goodwin, C.R.; McLean, J.A.; Li, D.; Webb, D.J. Glia co-culture with neurons in microfluidic platforms promotes the formation and stabilization of synaptic contacts. Lab Chip 2013, 13, 3008–3021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qaseem, A.; Snow, V.; Cross, J.T., Jr.; Forciea, M.A.; Hopkins, R., Jr.; Shekelle, P.; Adelman, A.; Mehr, D.; Schellhase, K.; Campos-Outcalt, D.; et al. American College of Physicians/American Academy of Family Physicians Panel on Dementia. Current pharmacologic treatment of dementia, a clinical practice guideline from the American College of Physicians and the American Academy of Family Physicians. Ann. Intern. Med. 2008, 148, 370–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidoryk-Węgrzynowicz, M.; Węgrzynowicz, M.; Lee, E.; Bowman, A.B.; Aschner, M. Role of astrocytes in brain function and disease. Toxicol. Pathol. 2011, 39, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Gutowicz, M. The influence of reactive oxygen species on the central nervous system. Postepy Hig. Med. Dosw. 2011, 65, 104–113. [Google Scholar] [CrossRef]
- Wawrzyniak-Gacek, A.; Jaworska-Adamu, J. Morphological central gray matter oligodendrocytes in old rats. Med. Weter. 2013, 69, 47–51. [Google Scholar]
- Philips, T.; Rothstein, J.D. Oligodendroglia, metabolic supporters of neurons. J. Clin. Investig. 2017, 127, 3271–3280. [Google Scholar] [CrossRef]
- Winiarska-Mieczan, A. Protective effect of tea against lead and cadmium-induced oxidative stress—A review. Biometals 2018, 31, 909–926. [Google Scholar] [CrossRef] [Green Version]
- Sajjad, R.; Arif, R.; Shah, A.A.; Manzoor, I.; Mustafa, G. Pathogenesis of Alzheimer’s disease, role of amyloid-β and hyperphosphorylated Tau protein. Indian J. Med. Microbiol. 2018, 80, 581–591. [Google Scholar] [CrossRef]
- Kubis, A.M.; Janusz, M. Alzheimer’s disease: New prospects in therapy and applied experimental models. Postepy Hig. Med. Dosw. 2008, 62, 372–392. [Google Scholar]
- Szwajgier, D. Anticholinesterase activity of selected phenolic acids and flavonoids—Interaction testing in model solutions. Ann. Agric. Environ. Med. 2015, 22, 690–694. [Google Scholar] [CrossRef] [PubMed]
- Shigemoto-Mogami, Y.; Hoshikawa, K.; Sato, K. Activated microglia disrupt the blood-brain barrier and induce chemokines and cytokines in a rat in vitro model. Front. Cell. Neurosci. 2018, 12, 494. [Google Scholar] [CrossRef] [PubMed]
- Gella, A.; Durany, N. Oxidative stress in Alzheimer disease. Cell Adhes. Migr. 2009, 3, 88–93. [Google Scholar] [CrossRef] [Green Version]
- Mariani, E.; Polidori, M.C.; Cherubini, A.; Mecocci, P. Oxidative stress in brain aging; neurodegenerative and vascular disease, an overview. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2005, 827, 65–75. [Google Scholar] [CrossRef]
- Park, J.S.; Davis, R.L.; Sue, C.M. Mitochondrial dysfunction in Parkinson’s disease, new mechanistic insights and therapeutic perspectives. Curr. Neurol. Neurosci. Rep. 2018, 18, 21. [Google Scholar] [CrossRef] [Green Version]
- Xicoy, H.; Wieringa, B.; Martens, G.J.M. The role of lipids in Parkinson’s disease. Cells 2019, 8, 27. [Google Scholar] [CrossRef] [Green Version]
- Saikia, A.; Bhattacharya, P.; Paul, S. Importance of dopamine in Parkinson’s disease. Adv. Tissue Eng. Regen. Med. 2018, 4, 47–48. [Google Scholar]
- Sołtan, W.; Gołębiewska, E.; Limon, J. Huntington disease—Three points of view. Forum Med. Rodz. 2011, 5, 108–114. [Google Scholar]
- Kumar, A.; Ratan, R.R. Oxidative stress and Huntington’s disease, the good; the bad; and the ugly. J. Huntingt. Dis. 2016, 5, 217–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galganska, H.; Karachitos, A.; Wojtkowska, M.; Stobienia, O.; Budzinska, M.; Kmita, H. Communication between mitochondria and nucleus, Putative role for VDAC in reduction/oxidation mechanism. Biochim. Biophys. Acta 2010, 1797, 1276–1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, K.G.; Banker, G.; Bourdette, D.; Forte, M. Axonal degeneration in multiple sclerosis, the mitochondrial hypothesis. Curr. Neurol. Neurosci. Rep. 2009, 9, 411–4117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haines, J.D.; Inglese, M.; Casaccia, P. Axonal damage in multiple sclerosis. Mt. Sinai J. Med. 2011, 78, 231–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurney, M.E.; Liu, R.; Althaus, J.S.; Hall, E.D.; Becker, D.A. Mutant CuZn superoxide dismutase in motor neuron disease. J. Inherit. Metab. Dis. 1998, 21, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Parakh, S.; Spencer, D.M.; Halloran, M.A.; Soo, K.Y.; Atkin, J.D. Redox regulation in amyotrophic lateral sclerosis. Oxid. Med. Cell. Longev. 2013, 2013, 408681. [Google Scholar] [CrossRef] [Green Version]
- Baumer, D.; Talbot, K.; Turner, M.R. Advances in motor neurone disease. J. R. Soc. Med. 2014, 107, 14–21. [Google Scholar] [CrossRef] [Green Version]
- Giridharan, S.; Srinivasan, M. Mechanisms of NF-κB p65 and strategies for therapeutic manipulation. J. Inflamm. Res. 2018, 11, 407–419. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.D.; Elias, R.J. The antioxidant and pro-oxidant activities of green tea polyphenols, a role in cancer prevention. Arch. Biochem. Biophys. 2010, 501, 65–72. [Google Scholar] [CrossRef] [Green Version]
- Andreoli, T.E. Free radicals and oxidative stress. Am. J. Med. 2000, 108, 650–651. [Google Scholar] [CrossRef]
- Barnham, K.J.; Masters, C.I.; Bush, A.I. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 2004, 3, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Saiki, C.; Okamura, H. Oxidative stress-tolerant stem cells from human exfoliated deciduous teeth decrease hydrogen peroxide-induced damage in organotypic brain slice cultures from adult mice. Int. J. Mol. Sci. 2019, 20, 1858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, L.; Cai, Q.; Wei, H. Alterations of antioxidant enzymes and oxidative damage to macromolecules in different organs of rats during aging. Free Radic. Biol. Med. 1998, 24, 1477–1484. [Google Scholar] [CrossRef]
- Pejić, S.; Stojiljković, A.; Todorović, A.; Gavrilović, L.; Pavlović, I.; Popović, N.; Pajović, S.Z. Antioxidant enzymes in brain cortex of rats exposed to acute, chronic and combined stress. Folia Biol. 2013, 64, 189–195. [Google Scholar] [CrossRef]
- Winiarska-Mieczan, A. The potential protective effect of green; black; red and white tea infusions against adverse effect of cadmium and lead during chronic exposure—A rat model study. Regul. Toxicol. Pharmacol. 2015, 73, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Schad, A.; Fahimi, H.D.; Völkl, A.; Baumgart, E. Expression of catalase mRNA and protein in adult rat brain, detection by nonradioactive in situ hybridization with signal amplification by catalyzed reporter deposition (ISH-CARD) and immunohistochemistry (IHC)/immunofluorescence (IF). J. Histochem. Cytochem. 2003, 51, 751–760. [Google Scholar] [CrossRef] [Green Version]
- Nishikawa, M.; Hashida, M.; Takakura, Y. Catalase delivery for inhibiting ROS-mediated tissue injury and tumor metastasis. Adv. Drug Deliv. Rev. 2009, 61, 319–326. [Google Scholar] [CrossRef]
- Ścibior, D.; Czeczot, H. Catalase, structure; properties; functions. Postepy Hig. Med. Dosw. 2006, 60, 170–180. [Google Scholar]
- Klomsiri, C.; Karplus, P.A.; Poole, L.B. Cysteine-based redox switches in enzymes. Antioxid. Redox Signal. 2011, 14, 1065–1077. [Google Scholar] [CrossRef] [Green Version]
- Lambe, A.T.; Krechmer, J.E.; Peng, Z.; Casar, J.R.; Carrasquillo, A.J.; Raff, J.D.; Jimenez, J.L.; Worsnop, D.R. HOx and NOx production in oxidation flow reactors via photolysis of isopropyl nitrite; isopropyl nitrite-d7; and 1;3-propyl dinitrite at λ = 254; 350; and 369 nm. Atmos. Meas. Tech. 2019, 12, 299–311. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Li, W.; Xu, Y.; Jin, E.; Tu, Y. Evaluation of the antioxidant effects of four main theaflavin derivatives through chemiluminescence and DNA damage analyses. Zhejiang Univ.-Sci. B 2011, 12, 744–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.Y.; Ke, D.S.; Chen, J.Y. Essential fatty acids and human brain. Acta Neurol. Taiwan 2009, 18, 231–241. [Google Scholar] [PubMed]
- Drechsel, D.A.; Estévez, A.G.; Barbeito, L.; Beckman, J.S. Nitric oxide-mediated oxidative damage and the progressive demise of motor neurons in ALS. Neurotox. Res. 2012, 22, 251–264. [Google Scholar] [CrossRef] [Green Version]
- Ahola, T.; Fellman, V.; Kjellmer, I.; Raivio, K.O.; Lapatto, R. Plasma 8-isoprostane is increased in preterm infants who develop bronchopulmonary dysplasia or periventricular leukomalacia. Pediatr. Res. 2004, 56, 88–93. [Google Scholar] [CrossRef] [Green Version]
- Yen, G.C.; Hsieh, C.L. Antioxidant effects of dopamine and related compounds. Biosci. Biotechnol. Biochem. 1997, 61, 1646–1649. [Google Scholar] [CrossRef] [Green Version]
- Choudhury, A.; Sahu, T.; Ramanujam, P.L.; Banerjee, A.K.; Chakraborty, I.; Kumar, A.; Arora, N. Neurochemicals; behaviours and psychiatric perspectives of neurological diseases. Neuropsychiatry 2018, 8, 395–424. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef]
- Salehi, F.; Behboudi, H.; Kavoosi, G.; Ardestani, S.K. Oxidative DNA damage induced by ROS-modulating agents with the ability to target DNA: A comparison of the biological characteristics of citrus pectin and apple pectin. Sci. Rep. 2018, 8, 13902. [Google Scholar] [CrossRef]
- Sharma, V.; Collins, L.B.; Chen, T.H.; Herr, N.; Takeda, S.; Sun, W.; Swenberg, J.A.; Nakamura, J. Oxidative stress at low levels can induce clustered DNA lesions leading to NHEJ mediated mutations. Oncotarget 2016, 7, 25377–25390. [Google Scholar] [CrossRef]
- Cenini, G.; Lloret, A.; Cascella, R. Oxidative stress in neurodegenerative diseases, from a mitochondrial point of view. Oxid. Med. Cell. Longev. 2019, 2019, 2105607. [Google Scholar] [CrossRef] [Green Version]
- Valavanidis, A.; Vlachogianni, T.; Fiotakis, C. 8-hydroxy-2′-deoxyguanosine (8-OHdG), A critical biomarker of oxidative stress and carcinogenesis. J. Environ. Sci. Health C 2009, 27, 120–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mecocci, P.; MacGarvey, U.; Kaufman, A.E.; Koontz, D.; Shoffner, J.M.; Wallace, D.C.; Beal, M.F. Oxidative damage to mitochondrial DNA shows marked age-dependent increases in human brain. Ann. Neurol. 1993, 34, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Shenkar, R.; Navidi, W.; Tavare, S.; Dang, M.H.; Chomyn, A.; Attardi, G.; Cortopassi, G.; Arnheim, N. The mutation rate of the human mtDNA deletion mtDNA4977. Am. J. Hum. Genet. 1996, 59, 772–780. [Google Scholar] [PubMed]
- Rivera-Mancía, S.; Pérez-Neri, I.; Ríos, C.; Tristán-López, L.; Rivera-Espinosa, L.; Montes, S. The transition metals copper and iron in neurodegenerative diseases. Chem. Biol. Interact. 2010, 186, 184–199. [Google Scholar] [CrossRef]
- Kanti Das, T.; Wati, M.R.; Fatima-Shad, K. Oxidative stress gated by Fenton and Haber Weiss reactions and its association with Alzheimer’s disease. Arch. Neurosci. 2015, 2, e60038. [Google Scholar] [CrossRef] [Green Version]
- Rae, T.D.; Schmidt, P.J.; Pufahl, R.A.; Culotta, V.C.; O’Halloran, T.V. Undetectable intracellular free copper, the requirement of a copper chaperone for superoxide dismutase. Science 1999, 284, 805–808. [Google Scholar] [CrossRef] [Green Version]
- Golub, M.S.; Germann, S.L.; Araiza, R.S.; Reader, J.R.; Griffey, S.M.; Lloyd, K.C. Movement disorders in the Hfe knockout mouse. Nutr. Neurosci. 2005, 8, 239–244. [Google Scholar] [CrossRef]
- Styś, A.; Starzyński, R.P.; Lipiński, P. The role of iron regulatory proteins in the control of iron metabolism in mammals. Biotechnologia 2011, 92, 66–75. [Google Scholar] [CrossRef] [Green Version]
- Georgieff, M.K. The role of iron in neurodevelopment: Fetal iron deficiency and the developing hippocampus. Biochem. Soc. Trans. 2008, 36, 1267–1271. [Google Scholar] [CrossRef] [Green Version]
- Stohs, S.J.; Bagchi, D. Oxidative mechanisms in the toxicity of metal ions. Free Radic. Biol. Med. 1995, 18, 321–336. [Google Scholar] [CrossRef] [Green Version]
- Chlebda, E.; Antonowicz-Juchniewicz, J.; Andrzejak, R. The effect of occupational exposure to heavy metals and arsenic on serum concentrations of carotenoids in copper foundry workers. Med. Pr. 2004, 55, 389–401. [Google Scholar] [PubMed]
- Hamed, E.A.; Meki, A.R.M.A.; Abd El-Mottaleb, N.A. Protective effect of green tea on lead-induced oxidative damage in rat’s blood and brain tissue homogenates. J. Physiol. Biochem. 2010, 66, 143–151. [Google Scholar] [CrossRef]
- Mao, T.; Han, C.; Wei, B.; Zhao, L.; Zhang, Q.; Deng, R.; Liu, J.; Luo, Y.; Zhang, Y. Protective effects of quercetin against cadmium chloride-induced oxidative injury in goat sperm and zygotes. Biol. Trace Elem. Res. 2018, 185, 344–355. [Google Scholar] [CrossRef]
- Czeczot, H.; Ścibior-Bentkowska, D.; Skrzycki, M.; Podsiad, M.; Karlik, W.; Bąkała, A.; Grono, D.; Wiechetek, M. Effect of cadmium on the activity of antioxidant enzymes in isolated rat hepatocytes. Med. Weter. 2009, 65, 55–60. [Google Scholar]
- Nemmiche, S. Oxidative signaling response to cadmium exposure. Toxicol. Sci. 2017, 156, 4–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winiarska-Mieczan, A. Protective effect of tannic acid on the brain of adult rats exposed to cadmium and lead. Environ. Toxicol. Pharmacol. 2013, 36, 9–18. [Google Scholar] [CrossRef]
- Gurer, H.; Ercal, N. Can antioxidants be beneficial in the treatment of lead poisoning? Free Radic. Biol. Med. 2000, 29, 927–945. [Google Scholar] [CrossRef]
- Patra, R.C.; Rautray, A.K.; Swarup, D. Oxidative stress in lead and cadmium toxicity and its amelioration. Vet. Med. Int. 2011, 2011, 457327. [Google Scholar] [CrossRef] [Green Version]
- Sandhir, R.; Julka, D.; Gill, K.D. Lipoperoxidative damage on lead exposure in rat brain and its implications on membrane bound enzymes. Pharmacol. Toxicol. 1994, 74, 66–71. [Google Scholar] [CrossRef]
- Sainath, S.B.; Meena, R.; Supriya, C.; Pratap Reddy, K.; Sreenivasula, R.P. Protective role of Centella asiatica on lead-induced oxidative stress and suppressed reproductive health in male rats. Environ. Toxicol. Pharmacol. 2011, 32, 146–154. [Google Scholar] [CrossRef]
- Casalino, E.; Calzaretti, G.; Sblano, C.; Landriscina, C. Molecular inhibitory mechanism of antioxidant enzymes in rat liver and kidney by cadmium. Toxicology 2002, 17, 37–50. [Google Scholar] [CrossRef]
- Bors, W.; Czapski, G.; Saran, M. An expanded function for superoxide dismutase. Free Radic. Res. Commun. 1991, 12, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Nelson, S.A.; Bose, S.K.; Grunwald, G.K.; Myhill, P.; McCord, J.M. The induction of human superoxide dismutase and catalase in vivo: A fundamentally new approach to antioxidant therapy. Free Rad. Biol. Med. 2006, 40, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Haouem, S.; El Hani, A. Effect of cadmium on lipid peroxidation and on some antioxidants in the liver, kidneys and testes of rats given diet containing cadmium-polluted radish bulbs. J. Toxicol. Pathol. 2013, 26, 359–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shagirtha, K.; Muthumani, M.; Prabu, S.M. Melatonin abrogates cadmium induced oxidative stress related neurotoxicity in rats. Eur. Rev. Med. Pharmacol. Sci. 2011, 15, 1039–1050. [Google Scholar] [PubMed]
- Gueroui, M.; Kechrid, Z. Evaluation of some biochemical parameters and brain oxidative stress in experimental rats exposed chronically to silver nitrate and the protective role of vitamin E and selenium. Toxicol. Res. 2016, 32, 301–309. [Google Scholar] [CrossRef] [Green Version]
- Renugadevi, J.; Prabu, S.M.; Hupathy, S.S. Protective role of a-tocopherol and ascorbic acid against cadmium induced neurotoxicity in rats. Int. J. Med. Sci. 2009, 2, 11–17. [Google Scholar]
- Pace, C.; Dagda, R.; Angermann, J. Antioxidants protect against arsenic induced mitochondrial cardio-toxicity. Toxics 2017, 5, 38. [Google Scholar] [CrossRef] [Green Version]
- Lohan, S.B.; Vitt, K.; Scholz, P.; Keck, C.M.; Meinke, M.C. ROS production and glutathione response in keratinocytes after application of β-carotene and VIS/NIR irradiation. Chem. Biol. Interact. 2018, 280, 1–7. [Google Scholar] [CrossRef]
- Chandravanshi, L.P.; Gupta, R.; Shukla, R.K. Arsenic-induced neurotoxicity by dysfunctioning cholinergic and dopaminergic system in brain of developing rats. Biol. Trace Elem. Res. 2019, 189, 118–133. [Google Scholar] [CrossRef]
- Dyatlov, V.A.; Dyatlova, O.M.; Parsons, P.J.; Lawrence, D.A.; Carpenter, D.O. Lipopolysaccharide and interleukin-6 enhance lead entry into cerebellar neurons, application of a new and sensitive flow cytometric technique to measure intracellular lead and calcium concentrations. Neurotoxicology 1998, 19, 293–302. [Google Scholar]
- Al-Oud, S.S. Heavy metal contents in tea and herb leaves. Pak. J. Biol. Sci. 2003, 6, 208–212. [Google Scholar]
- El-Sayed, I.H.; Lotfy, M.; El-Khawaga, O.A.Y.; Nasif, W.A.; El-Shahat, M. Prominent free radicals scavenging activity of tannic acid in lead-induced oxidative stress in experimental mice. Toxicol. Ind. Health 2006, 4, 157–163. [Google Scholar] [CrossRef]
- Lopes, G.K.B.; Schulman, H.M.; Hermes-Lima, M. Polyphenol tannic acid inhibits hydroxyl radical formation from Fenton reaction by complexing ferrous ions. Biochim. Biophys. Acta 1999, 1472, 142–152. [Google Scholar] [CrossRef]
- Marković, Z. Study of the mechanisms of antioxidative action of different antioxidants. J. Serb. Soc. Comput. Mech. 2016, 10, 135–150. [Google Scholar] [CrossRef]
- Leopoldini, M.; Russo, N.; Toscano, M. The molecular basis of working mechanism of natural polyphenolic antioxidants. Food Chem. 2011, 125, 288–306. [Google Scholar] [CrossRef]
- Woldu, A.S.; Mai, J. Computation of the bond dissociation enthalpies and free energies of hydroxylic antioxidants using the ab initio Hartree–Fock method. Redox Rep. 2013, 17, 252–274. [Google Scholar] [CrossRef] [PubMed]
- Migliore, A.; Polizzi, N.F.; Therien, M.J.; Beratan, D.N. Biochemistry and theory of proton-coupled electron transfer. Chem. Rev. 2014, 114, 3381–3465. [Google Scholar] [CrossRef] [PubMed]
- Dalle-Donne, I.; Milzani, A.; Gagliano, N.; Colombo, R.; Giustarini, D.; Rossi, R. Molecular mechanisms and potential clinical significance of S-glutathionylation. Antioxid. Redox Signal. 2008, 10, 445–473. [Google Scholar] [CrossRef]
- Adi, P.J.; Burra, S.P.; Vataparti, A.R.; Matcha, B. Calcium; zinc and vitamin E ameliorate cadmium-induced renal oxidative damage in albino Wistar rats. Toxicol. Rep. 2016, 3, 591–597. [Google Scholar] [CrossRef] [Green Version]
- Ashafaq, M.; Tabassum, H.; Vishnoi, S.; Salman, M.; Raisuddin, S.; Parvez, S. Tannic acid alleviates lead acetate-induced neurochemical perturbations in rat brain. Neurosci. Lett. 2016, 617, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, T.; Abdulla, S.; Yakulasamy, V.; Selvaraj, M.; Mathan, R. A mechanism underlying the neurotoxicity induced by sodium fluoride and its reversal by epigallocatechin gallate in the rat hippocampus, involvement of NrF2/Keap-1signaling pathway. J. Basic Appl. Zool. 2018, 79, 17. [Google Scholar] [CrossRef] [Green Version]
- Kom, H.H.; Nageshwar, M.; Srilatha, K.; Reddy, K.P. Protective effect of quercetin on weight drop injury model-induced neuroinflammation alterations in brain of mice. J. Apple. Pharm. Sci. 2019, 9, 96–103. [Google Scholar]
- Unsal, C.; Kanter, M.; Aktas, C.; Erboga, M. Role of quercetin in cadmium-induced oxidative stress, neuronal damage, and apoptosis in rats. Toxicol. Ind. Health 2015, 31, 1106–1115. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.S.; Wang, J.L.; Feng, D.Y.; Qin, H.Z.; Wen, H.; Yin, Z.M.; Gao, G.D.; Li, C. Protective effect of quercetin against oxidative stress and brain edema in an experimental rat model of subarachnoid hemorrhage. Int. J. Med. Sci. 2014, 11, 282–290. [Google Scholar] [CrossRef] [Green Version]
- Zaki, N.I.; Hassanin, Z.A.; Sabry, D. Evaluation of the protective role of quercetin and lecithin on Ifosfamide neurotoxicity in rats. Internat. J. Adv. Res. Biol. Sci. 2016, 11, 9–23. [Google Scholar] [CrossRef]
- Ghahremani, S.; Soodi, M.; Atashi, A. Quercetin ameliorates chlorpyrifos-induced oxidative stress in the rat brain: Possible involvement of PON2 pathway. J. Food Biochem. 2018, 42, e12530. [Google Scholar] [CrossRef]
- Malekiyan, R.; Abdanipour, A.; Sohrabi, D.; Anarkooli, I.J. Antioxidant and neuroprotective effects of lycopene and insulin in the hippocampus of streptozotocin-induced diabetic rats. Biomed. Rep. 2019, 10, 47–54. [Google Scholar] [CrossRef]
- Wang, J.; Li, L.; Wang, Z.; Cui, Y.; Tan, X.; Yuan, T.; Liu, Q.; Liu, Z.; Liu, X. Supplementation of lycopene attenuates lipopolysaccharide-induced amyloidogenesis and cognitive impairments via mediating neuroinflammation and oxidative stress. J. Nutr. Biochem. 2018, 56, 16–25. [Google Scholar] [CrossRef]
- Zhao, B.; Ren, B.; Guo, R.; Zhang, W.; Ma, S.; Yao, Y.; Yuan, T.; Liu, Z.; Liu, X. Supplementation of lycopene attenuates oxidative stress induced neuroinflammation and cognitive impairment via Nrf2/NF-κB transcriptional pathway. Food Chem. Toxicol. 2017, 109, 505–516. [Google Scholar] [CrossRef]
- Flora, G.; Gupta, D.; Tiwari, A. Preventive efficacy of bulk and nanocurcumin against lead-induced oxidative stress in mice. Biol. Trace Elem. Res. 2013, 152, 31–40. [Google Scholar] [CrossRef] [PubMed]
- García-Niño, W.R.; Zatarain-Barrón, Z.L.; Hernández-Pando, R.; Vega-García, C.C.; Tapia, E.; Pedraza-Chaverri, J. Oxidative stress markers and histological analysis in diverse organs from rats treated with a hepatotoxic dose of Cr(VI): Effect of curcumin. Biol. Trace Elem. Res. 2015, 167, 130–145. [Google Scholar] [CrossRef] [PubMed]
- Hegazi, M.M.; Mostafa, H.A.; Hegazi, M.A.M.; Elsaid, A.S.I. Effect of green tea and curcumin on the oxidative stress caused by gasoline in male mice brain. Egypt. J. Exp. Biol. 2009, 5, 473–480. [Google Scholar]
- El-Sokkary, G.H.; Awadalla, E.A. The protective role of vitamin C against cerebral and pulmonary damage induced by cadmium chloride in male adult albino rat. Open Neuroendocrinol. J. 2011, 4, 1–8. [Google Scholar] [CrossRef]
- Galal, M.K.; Khalaf, A.A.A.; Ogaly, H.A.; Ibrahim, M.A. Vitamin E attenuates neurotoxicity induced by deltamethrin in rats. BMC Complement. Altern. Med. 2014, 14, 458. [Google Scholar] [CrossRef] [Green Version]
- Alzoubi, K.H.; Halboup, A.M.; Alomari, M.A.; Khabour, O.F. The neuroprotective effect of vitamin E on waterpipe tobacco smoking-induced memory impairment: The antioxidative role. Life Sci. 2019, 222, 46–52. [Google Scholar] [CrossRef]
- Amara, I.B.; Soudani, N.; Hakim, A.; Troudi, A.; Zeghal, K.M.; Boudawara, T.; Zeghal, N. Selenium and vitamin E; natural antioxidants; protect rat cerebral cortex against dimethoate-induced neurotoxicity. Pestic. Biochem. Physiol. 2011, 101, 165–174. [Google Scholar] [CrossRef]
- Beytut, E.; Yilmaz, S.; Aksakal, M.; Polat, S. The possible protective effects of vitamin E and selenium administration in oxidative stress caused by high doses of glucocorticoid administration in the brain of rats. J. Trace Elem. Med. Biol. 2018, 45, 131–135. [Google Scholar] [CrossRef]
- Al-Olayan, E.M.; El-Khadragy, M.F.; Abdel Moneim, A.E. The protective properties of melatonin against aluminium-induced neuronal injury. Int. J. Exp. Path. 2015, 96, 196–202. [Google Scholar] [CrossRef]
- Ma, S.H.; Zhang, L.L.; Jiang, Q.Q. Protective effect of bioflavonoid morin on cadmium induced oxidative neuropathy. BioMed Res. 2017, 28, 1148–1154. [Google Scholar]
- Subash, S.; Subramanian, P. Morin a flavonoid exerts antioxidant potential in chronic hyperammonemic rats: A biochemical and histopathological study. Mol. Cell. Biochem. 2009, 327, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, N.A.; Abdel Gawad, H.S. Taurine dietary supplementation attenuates brain; thyroid; testicular disturbances and oxidative stress in streptozotocin-induced diabetes mellitus in male rats. Beni-Suef Univ. J. Basic Appl. Sci. 2017, 6, 247–252. [Google Scholar] [CrossRef]
- Niu, X.; Zheng, S.; Liu, H.; Li, S. Protective effects of taurine against inflammation; apoptosis; and oxidative stress in brain injury. Mol. Med. Rep. 2018, 18, 4516–4522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akkoyun, H.T.; Bengü, A.Ş.; Ulucan, A.; Akkoyun, M.B.; Ekin, S.; Temel, Y.; Çiftçi, M. Effect of astaxanthin on rat brains against oxidative stress induced by cadmium: Biochemical, histopathological evaluation. J. Instit. Sci. Technol. 2018, 8, 33–39. [Google Scholar] [CrossRef] [Green Version]
- El-Agamy, S.E.; Abdel-Aziz, E.K.; Wahdan, S.; Esmat, A.; Azab, S.S. Astaxanthin ameliorates doxorubicin-induced cognitive impairment (chemobrain) in experimental rat model, impact on oxidative; inflammatory; and apoptotic machineries. Mol. Neurobiol. 2018, 55, 5727–5740. [Google Scholar] [CrossRef] [PubMed]
- Loh, K.P.; Qi, J.; Tan, B.K.; Liu, X.H.; Wei, B.G.; Zhu, Y.Z. Leonurine protects middle cerebral artery occluded rats through antioxidant effect and regulation of mitochondrial function. Stroke 2010, 41, 2661–2668. [Google Scholar] [CrossRef] [Green Version]
- Samarghandian, S.; Farkhondeh, T.; Samini, F.; Borji, A. Protective effects of carvacrol against oxidative stress induced by chronic stress in rat’s brain; liver; and kidney. Biochem. Res. Int. 2016, 2016, 2645237. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.; Ikram, M.; Muhammad, T.; Park, J.; Kim, M.O. Caffeine modulates cadmium-induced oxidative stress, neuroinflammation, and cognitive impairments by regulating Nrf-2/HO-1 in vivo and in vitro. J. Clin. Med. 2019, 8, 680. [Google Scholar] [CrossRef] [Green Version]
- Nalagoni, C.S.R.; Karnati, P.R. Protective effect of resveratrol against neuronal damage through oxidative stress in cerebral hemisphere of aluminum and fluoride treated rats. Interdiscip. Toxicol. 2016, 9, 78–82. [Google Scholar] [CrossRef] [Green Version]
- Turkmen, R.; Birdane, Y.O.; Demirel, H.H.; Kabu, M.; Ince, S. Protective effects of resveratrol on biomarkers of oxidative stress; biochemical and histopathological changes induced by sub-chronic oral glyphosate-based herbicide in rats. Toxicol. Res. 2019, 8, 238–245. [Google Scholar] [CrossRef] [Green Version]
- Tan, B.L.; Norhaizan, M.E.; Liew, W.P.P.; Rahman, H.S. Antioxidant and oxidative stress: A mutual interplay in age-related diseases. Front. Pharmacol. 2018, 9, 1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacic, P.; Somanathan, R. Cell signaling and receptors with resorcinols and flavonoids, redox; reactive oxygen species, and physiological effects. J. Recept. Signal Transduct. 2011, 31, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.M.; Lin, J.K.; Liu, S.H.; Lin-Shiau, S.Y. Novel regimen through combination of memantine and tea polyphenol for neuroprotection against brain excitotoxicity. J. Neurosci. Res. 2008, 86, 2696–2704. [Google Scholar] [CrossRef] [PubMed]
- Ide, K.; Yamada, H.; Takuma, N.; Kawasaki, Y.; Harada, S.; Nakase, J.; Ukawa, Y.; Sagesaka, Y.M. Effects of green tea consumption on cognitive dysfunction in an elderly population, A randomized placebo-controlled study. Nutr. J. 2016, 15, 49. [Google Scholar] [CrossRef] [Green Version]
- Haque, A.M.; Hashimoto, M.; Katakura, M.; Hara, Y.; Shido, O. Green tea catechins prevent cognitive deficits caused by Aβ1–40 in rats. J. Nutr. Biochem. 2008, 19, 619–626. [Google Scholar] [CrossRef]
- Ide, K.; Matsuoka, N.; Yamada, H.; Furushima, D.; Kawakami, K. Effects of tea catechins on Alzheimer’s disease, recent updates and perspectives. Molecules 2018, 23, 2357. [Google Scholar] [CrossRef] [Green Version]
- Ward, R.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 2014, 13, 1045–1060. [Google Scholar] [CrossRef] [Green Version]
- Biasibetti, R.; Tramontina, A.C.; Costa, A.P.; Dutra, M.F.; Quincozes-Santos, A.; Nardin, P.; Bernardi, C.L.; Wartchow, K.M.; Lunardi, P.S.; Goncalves, C.A. Green tea (−)epigallocatechin-3-gallate reverses oxidative stress and reduces acetylcholinesterase activity in a streptozotocin-induced model of dementia. Behav. Brain Res. 2013, 236, 186–193. [Google Scholar] [CrossRef]
- Khalaf, A.A.; Moselhy, W.A.; Abdel-Hamed, M.I. The protective effect of green tea extract on lead induced oxidative and DNA damage on rat brain. Neurotoxicology 2012, 33, 280–289. [Google Scholar] [CrossRef]
- Meki, A.R.; Alghasham, A.; El-Deeb, E.S. Effect of green tea extract on lead toxicity in different organs of rats. Int. J. Health Sci. (Quassim) 2011, 5, 12–15. [Google Scholar]
- Moneim, A.E.A. Evaluating the potential role of pomegranate peel in aluminum-induced oxidative stress and histopathological alterations in brain of female rats. Biol. Trace Elem. Res. 2012, 150, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Elkhadragy, M.F.; Kassab, R.B.; Metwally, D.; Almeer, R.S.; Abdel-Gaber, R.; Al-Olayan, E.M.; Essawy, E.A.; Amin, H.K.; Abdel Moneim, A.E. Protective effects of Fragaria ananassa methanolic extract in a rat model of cadmium chloride-induced neurotoxicity. Biosci. Rep. 2018, 38, BSR20180861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guesmi, F.; Bellamine, H.; Landoulsi, A. Hydrogen peroxide-induced oxidative stress; acetylcholinesterase inhibition; and mediated brain injury attenuated by Thymus algeriensis. Appl. Physiol. Nutr. Metab. 2018, 43, 1275–1281. [Google Scholar] [CrossRef] [Green Version]
- Abdou, H.M.; Wahby, M.M. Neuroprotection of grape seed extract and pyridoxine against triton-induced neurotoxicity. Oxid. Med. Cell. Longev. 2016, 2016, 8679506. [Google Scholar] [CrossRef]
- Singh, S.; Kaur, S.; Budhiraja, R.D. Chlorpyrifos-induced oxidative stress in rat’s brain and protective effect of grape seed extract. J. Phytopharmacol. 2013, 2, 26–33. [Google Scholar]
- Elhadidy, M.E.; Sawie, H.G.; Meguid, N.A.; Khadrawy, Y.A. Protective effect of ashwagandha (Withania somnifera) against neurotoxicity induced by aluminum chloride in rats. Asian Pac. J. Trop. Biomed. 2018, 8, 59–66. [Google Scholar]
- El-Masry, T.A.; Emara, A.M.; El-Shitany, N.A. Possible protective effect of propolis against lead induced neurotoxicity in animal model. J. Evol. Biol. Res. 2011, 3, 4–11. [Google Scholar]
- Kakoolaki, S.; Talas, Z.S.; Cakir, O.; Ciftci, O.; Ozdemir, I. Role of propolis on oxidative stress in fish brain. Basic Clin. Neurosci. 2013, 4, 153–158. [Google Scholar]
- Mainak, M.; Mahammed, M.; Saheli, K.; Debjit, D.; Bhusan, C.S. Neuronal and oxidative damage in the catfish brain alleviated after Mucuna seed extract treatment. Int. J. Pharmacogn. Phytochem. Res. 2017, 9, 52–57. [Google Scholar] [CrossRef]
- Singh, T.; Kasture, S.B.; Mohanty, P.K.; Jaliwala, Y.; Karchuli, M.S.; Agarwal, A.; Yadav, Y. Cyclophosphamide-induced oxidative stress in brain, Protective effect of Garcinia indica fruit extract. Int. J. Pharm. Life Sci. 2011, 2, 1035–1040. [Google Scholar]
- Yun, H.J.; Kim, I.; Kwon, S.H.; Kang, J.S.; Om, A.S. Protective effect of Chlorella vulgaris against lead-induced oxidative stress in rat brains. J. Health Sci. 2011, 57, 245–254. [Google Scholar] [CrossRef] [Green Version]
- Vijaya, P.; Sharma, S. Protective effects of natural antioxidant supplementation on cadmium induced toxicity in albino mice. J. Innov. Pharmac. Biol. Sci. 2018, 5, 16–21. [Google Scholar]
- Oyenihi, O.R.; Afolabi, B.A.; Oyenihi, A.B.; Ogunmokun, O.J.; Oguntibeju, O.O. Hepato- and neuro-protective effects of watermelon juice on acute ethanol-induced oxidative stress in rats. Toxicol. Rep. 2016, 3, 288–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vora, S.R.; Patil, R.B.; Pillai, M.M. Protective effects of Petroselinum crispum (Mill) Nyman ex A. W. Hill leaf extract on D-galactose-induced oxidative stress in mouse brain. Indian J. Exp. Biol. 2009, 47, 338–342. [Google Scholar]
- Garavaglia, J.; Markoski, M.M.; Oliveira, A.; Marcadenti, A. Grape seed oil compounds, biological and chemical actions for health. Nutr. Metab. Insights 2016, 9, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Reddy, P.H.; Manczak, M.; Yin, X.; Grady, M.C.; Mitchell, A.; Tonk, S.; Kuruva, C.S.; Bhatti, J.S.; Kandimalla, R.; Vijayan, M.; et al. Protective effects of Indian spice curcumin against amyloid-β in Alzheimer’s disease. J. Alzheimers Dis. 2018, 61, 843–866. [Google Scholar] [CrossRef]
- Ozguner, F.; Armagan, A.; Koyu, A.; Calıskan, S.; Koylu, H. A novel antioxidant agent caffeic acid phenethyl ester (CAPE) prevents shock wave-induced renal tubular oxidative stress. Urol. Res. 2005, 33, 239–243. [Google Scholar] [CrossRef]
- Prasad, S.; Tyagi, A.K.; Aggarwal, B.B. Recent developments in delivery; bioavailability; absorption and metabolism of curcumin, the golden pigment from golden spice. Cancer Res. Treat. 2014, 46, 2–18. [Google Scholar] [CrossRef] [Green Version]
- Brondino, N.; Re, S.; Boldrini, A.; Cuccomarino, A.; Lanati, N.; Barale, F.; Politi, P. Curcumin as a therapeutic agent in dementia: A mini systematic review of human studies. Sci. World J. 2014, 2014, 174282. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.-H.; Loo, C.-Y.; Bebawy, M.; Luk, F.; Mason, R.S.; Rohanizadeh, R. Curcumin and its derivatives, their application in neuropharmacology and neuroscience in the 21st century. Curr. Neuropharmacol. 2013, 11, 338–378. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, A.E.; de Oliveira, P.A.; de Souza, L.F.; da Silva, D.G.H.; Danielski, S.; Santos, D.B.; de Almeida, E.A.; Prediger, R.D.; Fisher, A.; Farina, M.; et al. Interaction of curcumin with manganese may compromise metal and neurotransmitter homeostasis in the hippocampus of young mice. Biol. Trace Elem. Res. 2014, 158, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Singh, J.P.; Kaur, A.; Singh, N. Phenolic compounds as beneficial phytochemicals in pomegranate (Punica granatum L.) peel: A Review. Food Chem. 2018, 261, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Zeghad, N.; Ahmed, E.; Belkhiri, A.; Heyden, Y.V.; Demeyer, K. Antioxidant activity of Vitis vinifera; Punica granatum; Citrus aurantium and Opuntia ficus indica fruits cultivated in Algeria. Heliyon 2019, 5, e01575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aragon, C.M.G.; Amit, Z.; Stotland, L.M. Studies on ethanol-brain catalase interaction, evidence for central ethanol oxidation. Alcohol. Clin. Exp. Res. 1991, 15, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Pavel, M.; Ristić, M.; Stević, T. Essential oils of Thymus pulegioides and Thymus glabrescens from Romania: Chemical composition and antimicrobial activity. J. Serb. Chem. Soc. 2010, 75, 27–34. [Google Scholar] [CrossRef]
- Sasikumar, V.; Subramaniam, A.; Aneesh, A.; Saravanan, G. Protective effect of alkaloids from Amaranthus Viridis Linn against hydrogen peroxide induced oxidative damage in human erythrocytes (RBC). Int. J. Clin. Endocrinol. Metab. 2015, 1, 49–53. [Google Scholar]
- Hussain, G.; Rasul, A.; Anwar, H.; Aziz, N.; Razzaq, A.; Wei, W.; Ali, M.; Li, J.; Li, X. Role of plant derived alkaloids and their mechanism in neurodegenerative disorders. Int. J. Biol. Sci. 2018, 14, 341–357. [Google Scholar] [CrossRef] [Green Version]
- Fried, N.T.; Elliott, M.B.; Oshinsky, M.L. The role of adenosine signaling in headache: A review. Brain Sci. 2017, 7, 30. [Google Scholar] [CrossRef] [Green Version]
- Gomes, C.V.; Kaster, M.P.; Tomé, A.R.; Agostinho, P.M.; Cunha, R.A. Adenosine receptors and brain diseases: Neuroprotection and neurodegeneration. Biochim. Biophys. Acta 2011, 1808, 1380–1399. [Google Scholar] [CrossRef] [Green Version]
- Krashia, P.; Nobili, A.; D’Amelio, M. Unifying hypothesis of dopamine neuron loss in neurodegenerative diseases: Focusing on Alzheimer’s disease. Front. Mol. Neurosci. 2019, 12, 123. [Google Scholar] [CrossRef]
- Czapski, G.A.; Szypuła, W.; Kudlik, M.; Wileńska, B.; Kania, M.; Danikiewicz, W.; Adamczyk, A. Assessment of antioxidative activity of alkaloids from Huperzia selago and Diphasiastrum complanatum using in vitro systems. Folia Neuropathol. 2014, 52, 394–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeida, M.B.; Costa-Malaquias, A.; Nascimento, J.L.M.; Oliveira, K.R.; Herculano, A.M.; Crespo-Lopez, M.E. Therapeutic concentration of morphine reduces oxidative stress in glioma cell line. Braz. J. Med. Biol. Res. 2014, 47, 398–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kannampalli, P.; Sengupta, J.N. Role of principal ionotropic and metabotropic receptors in visceral pain. J. Neurogastroenterol. Motil. 2015, 21, 147–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Simon, F.M.; Arenzana, F.J.; Rodriguez, R.E. In vivo effects of morphine on neuronal fate and opioid receptor expression in zebrafish embryos. Eur. J. Neurosci. 2010, 32, 550–559. [Google Scholar] [CrossRef]
- Crupi, R.; Impellizzeri, D.; Cuzzocrea, S. Role of metabotropic glutamate receptors in neurological disorders. Front. Mol. Neurosci. 2019, 12, 20. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Wang, Y.; Dong, Q.; Wu, S.; Xiao, X.; Hu, J.; Chai, Z.; Zhang, Y. Morphine protects against intracellular amyloid toxicity by inducing estradiol release and upregulation of Hsp70. J. Neurosci. 2011, 31, 16227–16240. [Google Scholar] [CrossRef] [Green Version]
- Forni, C.; Facchiano, F.; Bartoli, M.; Pieretti, S.; Facchiano, A.; D’Arcangelo, D.; Norelli, S.; Valle, G.; Nisini, R.; Beninati, S.; et al. Beneficial role of phytochemicals on oxidative stress and age-related diseases. BioMed Res. Int. 2019, 2019, 8748253. [Google Scholar] [CrossRef] [Green Version]
- Sirmali, R.; Ginis, Z.; Sirmali, M.; Solak, O.; Şeliman, B.; Ağaçkiran, Y.; Delibaş, N. Vitamin C as an antioxidant, evaluation of its role on pulmonary contusion experimental model. Turk. J. Med. Sci. 2014, 44, 905–913. [Google Scholar] [CrossRef]
- Salman, N.A.A.F.A.; El-Safty, F.E.-N.A.-H.; El-Habeby, M.M.; El-Kholy, W.B.; El-Akabawy, G.F.A. Vitamin C attenuates the toxic effect of nutmeg on primary visual occipital cortex in rats. Folia Morphol. 2019, 78, 33–38. [Google Scholar]
- Dortaj, H.; Yadegari, M.; Hosseini, S.A.M.; Abbasi, S.A.; Anvari, M. Stereological method for assessing the effect of vitamin C administration on the reduction of acrylamide-induced neurotoxicity. Basic Clin. Neurosci. 2018, 9, 27–34. [Google Scholar] [CrossRef]
- Raederstorff, D.; Wyss, A.; Calder, P.C.; Weber, P.; Eggersdorfer, M. Vitamin E function and requirements in relation to PUFA. Br. J. Nutr. 2015, 114, 1113–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salehi, I.; Saidijam, M.; Vahidinia, A.A.; Sohrabi, M.; Soleimani Asl, S. High fat diet induced neurotoxicity alters following vitamin E and C administration in hippocampus of male rats. Gene Cell Tissue 2017, 4, e58383. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Kim, H. Inhibitory effect of astaxanthin on oxidative stress-induced mitochondrial dysfunction—A mini-review. Nutrients 2018, 10, 1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambati, R.R.; Phang, S.M.; Ravi, S.; Aswathanarayana, R.G. Astaxanthin: Sources, extraction, stability, biological activities and its commercial applications—A review. Mar. Drugs 2014, 12, 128–152. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.J.; Chen, W.; Fu, X.T.; Ma, J.K.; Wang, M.H.; Hou, Y.J.; Tian, D.C.; Fu, X.Y.; Fan, C.D. Reversal of homocysteine-induced neurotoxicity in rat hippocampal neurons by astaxanthin, evidences for mitochondrial dysfunction and signaling crosstalk. Cell Death Dis. 2018, 4, 50. [Google Scholar] [CrossRef] [PubMed]
- Kaya, E.; Yilmaz, S.; Çeribaşi, A.O.; Telo, S. Protective effect of lycopene on diethylnitrosamine-induced oxidative stress and catalase expression in rats. Ankara Üniv. Vet. Fak. Derg. 2019, 66, 43–52. [Google Scholar]
- Kelkel, M.; Schumacher, M.; Dicato, M.; Diederich, M. Antioxidant and anti-proliferative properties of lycopene. Free Radic. Res. 2011, 45, 925–940. [Google Scholar] [CrossRef]
- Crowe-White, K.M.; Phillips, T.A.; Ellis, A.C. Lycopene and cognitive function. J. Nutr. Sci. 2019, 8, e20. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.B.; Wang, R.; Yi, Y.F.; Gao, Z.; Chen, Y.Z. Lycopene mitigates β-amyloid induced inflammatory response and inhibits NF-κB signaling at the choroid plexus in early stages of Alzheimer’s disease rats. J. Nutr. Biochem. 2018, 53, 66–71. [Google Scholar] [CrossRef]
- Kuriyama, S.; Hozawa, A.; Ohmori, K.; Shimazu, T.; Matsui, T.; Ebihara, S.; Awata, S.; Nagatomi, R.; Arai, H.; Tsuji, I. Green tea consumption and cognitive function, A cross-sectional study from the Tsurugaya Project 1. Am. J. Clin. Nutr. 2006, 83, 355–361. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.J.; He, C.H.; Li, S.; Zhang, S.Y.; Duan, S.Y.; Sun, H.P.; Shen, Y.P.; Xu, Y.; Yin, J.Y.; Pan, C.W. Tea consumption is associated with cognitive impairment in older Chinese adults. Aging Ment. Health 2017, 22, 1232–1238. [Google Scholar] [CrossRef] [PubMed]
- Nurk, E.; Refsum, H.; Drevon, C.A.; Tell, G.S.; Nygaard, H.A.; Engedal, K.; Smith, A.D. Intake of flavonoid-rich wine; tea; and chocolate by elderly men and women is associated with better cognitive test performance. J. Nutr. 2009, 139, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Gwee, X.; Kua, E.H.; Ng, T.P. Cognitive function and tea consumption in community dwelling older Chinese in Singapore. J. Nutr. Health Aging 2010, 14, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Q.; Dong, B.R.; Zhang, Y.L.; Wu, H.M.; Liu, Q.X. Association of cognitive impairment with smoking; alcohol consumption; tea consumption; and exercise among Chinese nonagenarians/centenarians. Cogn. Behav. Neurol. 2009, 22, 190–196. [Google Scholar] [CrossRef]
- Arab, L.; Biggs, M.L.; O’Meara, E.S.; Longstreth, W.T.; Crane, P.K.; Fitzpatrick, A.L. Gender differences in tea, coffee, and cognitive decline in the elderly: The Cardiovascular Health Study. J. Alzheimers Dis. 2011, 27, 553–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, L.; Li, J.; Ng, T.P.; Lee, T.S.; Kua, E.H.; Zeng, Y. Tea drinking and cognitive function in oldest-old Chinese. J. Nutr. Health Aging 2012, 16, 754–758. [Google Scholar] [CrossRef] [Green Version]
- Noguchi-Shinohara, M.; Yuki, S.; Dohmoto, C.; Ikeda, Y.; Samuraki, M.; Iwasa, K.; Yokogawa, M.; Asai, K.; Komai, K.; Nakamura, H.; et al. Consumption of green tea; but not black tea or coffee; is associated with reduced risk of cognitive decline. PLoS ONE 2014, 9, e96013. [Google Scholar] [CrossRef] [Green Version]
- Ng, T.P.; Feng, L.; Niti, M.; Kua, E.H.; Yap, K.B. Tea consumption and cognitive impairment and decline in older Chinese adults. Am. J. Clin. Nutr. 2008, 88, 224–231. [Google Scholar] [CrossRef]
- Hosseini, T.N.; Babakhani, B.; Hosseini, T.A.; Vahabi, Z.; Soltanzadeh, A. Non-genetic factors associated with the risk of Parkinson’s disease in Iranian patients. Funct. Neurol. 2013, 28, 107–113. [Google Scholar]
- Larsson, S.C.; Orsini, N. Coffee consumption and risk of dementia and Alzheimer’s disease: A dose-response meta-analysis of prospective studies. Nutrients 2018, 10, 1501. [Google Scholar] [CrossRef] [Green Version]
- Eskelinen, M.H.; Ngandu, T.; Tuomilehto, J.; Soininen, H.; Kivipelto, M. Midlife coffee and tea drinking and the risk of late-lide dementia, a population-based CAIDE Study. J. Alzheimers Dis. 2009, 16, 85–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kataoka, Y.; Utsunomiya, K.; Kimbara, A.; Fukushima, K.; Mori, T.; Shiba, J. Preventive effect of green tea containing theanine at a high concentration on dementia in aged volunteers. J. Jpn. Mibyou. Syst. Assoc. 2009, 15, 17–23. [Google Scholar]
- Ide, K.; Yamada, H.; Takuma, N.; Park, M.; Wakamiya, N.; Nakase, J.; Ukawa, Y.; Sagesaka, Y.M. Green tea consumption affects cognitive dysfunction in the elderly, A pilot study. Nutrients 2014, 6, 4032–4042. [Google Scholar] [CrossRef] [PubMed]
- Park, S.K.; Jung, I.C.; Lee, W.K.; Lee, Y.S.; Park, H.K.; Go, H.J.; Kim, K.; Lim, N.K.; Hong, J.T.; Ly, S.Y.; et al. A combination of green tea extract and L-theanine improves memory and attention in subjects with mild cognitive impairment, A double-blind placebo-controlled study. J. Med. Food 2011, 14, 334–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Ding, D.; Wang, J.; Zhao, Q.; Meng, H.; Li, H.; Gao, Y.T.; Shu, X.O.; Tanner, C.M.; Hong, Z.; et al. Parkinson’s disease research in a prospective cohort in China. Parkinsonism Relat. Disord. 2015, 21, 1200–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, K.; Melo van Lent, D.; Wolfsgruber, S.; Weinhold, L.; Kleineidam, L.; Bickel, H.; Scherer, M.; Eisele, M.; van den Bussche, H.; Wiese, B.; et al. Prospective Associations between Single Foods; Alzheimer’s Dementia and Memory Decline in the Elderly. Nutrients 2018, 10, 852. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Cassidy, A.; Schwarzschild, M.A.; Rimm, E.B.; Ascherio, A. Habitual intake of dietary flavonoids and risk of Parkinson disease. Neurology 2012, 78, 1138–1145. [Google Scholar] [CrossRef] [Green Version]
- Rusek, M.; Pluta, R.; Ułamek-Kozioł, M.; Czuczwar, S.J. Ketogenic diet in Alzheimer’s disease. Int. J. Mol. Sci. 2019, 20, 3892. [Google Scholar] [CrossRef] [Green Version]
- Włodarek, D. Role of ketogenic diets in neurodegenerative diseases (Alzheimer’s disease and Parkinson’s disease). Nutrients 2019, 11, 169. [Google Scholar] [CrossRef] [Green Version]
- Milder, J.; Patel, M. Modulation of oxidative stress and mitochondrial function by the ketogenic diet. Epilepsy Res. 2012, 100, 295–303. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Cao, H.; Wen, J.; Xu, M. Green tea polyphenol (−)-epigallocatechin-3-gallate enhances the inhibitory effect of huperzine A on acetylcholinesterase by increasing the affinity with serum albumin. Nutr. Neurosci. 2009, 12, 142–148. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Protective Effect | Design | Animals | Target Sites | References | |
|---|---|---|---|---|---|
| Tannic acid | ↑ SOD after 12 weeks; ↑ CAT both after 6 and 12 weeks | 7 mg Cd (as cadmium chloride) and 50 mg Pb (as lead acetate) per kg of feed for 6 or 12 weeks; tannic acid with drink (0, 0.5, 1, 1.5, 2 or 2.5% solutions) for 6 or 12 weeks | Male Wistar rats | Total brain | [66] |
| ↑ SOD after 12 weeks; ↑ CAT both after 6 and 12 weeks | aqueous solutions of [Cd (7 or 14 mg L−1 distiller water) or Pb (50 or 100 mg L−1 distilled water)] or 2 % tannic acid solution, alternatively every 7 days, for 6 or 12 weeks | ||||
| Tannic acid | ↓ LPO; ↑ GSH; ↑ GST; ↑ GPX; ↑ SOD; ↑ CAT | 50 mg kg−1 bw lead acetate intraperitoneally three times a week for two weeks; 50 mg kg−1 bw tanic acid orally three times a week for two weeks | Male Wistar rats | Total brain | [91] |
| Epigallocatechin gallate (EGCG) | ↑ CAT; ↑ SOD; ↑ GPX; ↑ GSH; ↑ GST; ↑ GR; ↑ G6PD; ↑ TSH; ↓ ROS; ↓ TBARS; ↓ NO; ↓ PC; ↑ vitamin C | 25 mg kg−1 bw fluoride (as NaF)) per day by intragastric administration for 4 weeks; 40 mg kg−1 bw EGCG administrated 30 min before administration of NaF per day by intragastric administration for 4 weeks | Male Wistar rats | Hippocampus | [92] |
| Quercetin | ↓ LPO; ↑ CAT; ↑ SOD; ↑ GPX | Mice with traumatic brain injuries; 20 mg kg−1 bw quercetin through intraperitoneal injection for 7 days | Mice | Total brain | [93] |
| Quercetin | ↓ MDA; ↑ CAT; ↑ SOD; ↑ GPX | 1 mg Cd (as cadmium chloride) kg−1 bw per day by injection for 30 days; 15 mg quercetin kg−1 bw orally for 30 days | Male Sprague-Dawley rats | Total brain | [94] |
| Quercetin | ↓ MDA; ↑ SOD; ↑ GSH | Rats with brain damage after subarachnoid hemorrhage; 10 or 50 mg kg−1 bw quercetin administered intraperitoneally at 30 min, 12 h, and 24 h after the subarachnoid hemorrhage insult | Male Sprague-Dawley rats | Cerebral cortex | [95] |
| Quercetin | ↑ GSH; ↓ NO | 80 mg kg−1 ifosfamide intraperitoneally for 5 consecutive days; 50 mg kg−1 bw quercetin orally for 6 consecutive days | Adult female rats | Cortex, cerebellum, striatum, pons, thalamus, hypothalamus | [96] |
| Quercetin | ↑ CAT; ↓ MDA; ↑ GPX; ↑ total thiol; | 10 mg kg−1 chlorpyrifos orally once a day by gavage for 1 month, 30 min after administration of quercetin; 20 mg kg−1 quercetin orally once a day by gavage for 1 month | Male Sprague-Dawley rats | Total brain | [97] |
| Lycopene | ↓ MDA; ↑ TAC; ↓neuronal cell death | Diabetic rats; 4 mg kg−1 lycopene orally for 8 weeks | Male Wistar rats | Hippocampus | [98] |
| Lycopene | ↑ GSH; ↑ CAT; ↑ SOD | 0.25 mg kg−1 per day lipopolysaccharide by injection for 9 days; 0.03% lycopene mixed with standard diet for 5 weeks | Male C57BL/6J mice | Total brain | [99] |
| Lycopene | ↓ MDA; ↑ GSH; ↑ SOD; ↑ GPX | 150 mg kg−1 per day D-galactose by intraperitoneally injection for 8 weeks; 50 mg kg−1 bw lycopene per day mixed with standard diet for 8 weeks | CD-1 male mice | Hippocampus | [100] |
| Curcumin | ↑ GSH; ↓ TBARS | 25 mg kg−1 lead acetate orally for 2 weeks; alone and after 1 h treated orally either with curcumin (15 mg kg−1) or nanocurcumin (15 mg kg−1) for 2 weeks | Swiss albino mice | Total brain | [101] |
| Curcumin | ↑ GSH; ↑ SOD; ↑ GPX; ↑ GR; ↑ GST | 15 mg kg−1 bw potassium dichromate by a single intraperitoneal injection on 10 days; 400 mg kg−1 bw curcumin orally for 10 days | Male Wistar rats | Total brain | [102] |
| Curcumin | ↑ CAT; ↑ SOD; ↑ total thiol; ↓ TBARS; ↓ AOPP; ↓ PC | Gasoline inhalation—2 hours daily; 3% powdered curcumin roots in feed | Male mice CD1 strain | Total brain | [103] |
| Vitamin C | ↓ MDA; ↑ SOD; ↑ GSH | 5 mg kg−1 bw cadmium chloride injected subcutaneously every day for 49 days; 100 mg kg−1 vitamin C injected subcutaneously every day for 49 days 30 min. before Cd injection | Male Sprague-Dawley rats | Total brain | [104] |
| Vitamin C, vitamin E | ↓ LPO; ↑ AChE; ↑ SOD; ↑ CAT; ↑ GPX; ↑ GSH; ↑ ATPases | 5 mg kg−1 bw per day cadmium chloride orally for 284 days; 50 mg kg−1 bw per day vitamin C and vitamin E orally for 248 days | Rats | Total brain | [77] |
| Vitamin E | ↓ MDA; ↓ NO; ↑ TAC | 0.6 mg kg−1 bw deltamethrin taken once daily via oral gavage for 30 days; 200 mg kg−1 bw vitamin E taken once daily via oral gavage for 30 days | Male albino rats | Total brain | [105] |
| Vitamin E | ↑ CAT; ↑ GPX | Waterpipe tobacco smoke exposure for one-hour session per day for five days per week for 1 month; 100 mg kg−1 vitamin E once a day by oral gavage for 1 month | Adult Wistar rats | Hippocapus | [106] |
| Vitamin E, selenium | ↓ AOPP; ↑ vitamin C; improved the diminished activities of antioxidative enzymes and the levels of GSH | 0.2 g L−1 drinking water dimethoate; 100 mg kg−1 diet vitamin E; 0.5 mg kg−1 diet selenium | Adult Wistar rats | Cerebral cortex tissue | [107] |
| Vitamin E, selenium | ↓ TBARS; improved the diminished activities of antioxidative enzymes and the levels of GSH | 100 mg kg−1 bw prednisolone injected intramuscularly for 3 consecutive days; 20 mg DL-α-tocopheryl acetate and 0.3 mg sodium selenite for 30 days by oral route | Male Wistar rats | Total brain | [108] |
| Vitamin E, selenium | ↑ SOD; ↓ TBARS; ↑ GSH; ↑ GST; ↑ GR; ↑ vitamin E | 20 mg L−1 AgNO3 in drinking water; 400 mg kg−1 diet vitamin E and mg L−1 selenium in drinking water | Male Wistar rats | Total brain | [76] |
| Melatonin | ↑ AChE; ↓ TBARS; ↑ GSH; ↑ vitamin C; ↑ vitamin E; ↑ TSH | 5 mg kg−1 bw cadmium chloride orally for 4 weeks; melatonin (10 mg kg bw) in ethanol subcutaneously for 4 weeks; the injection of melatonin was 30 min before Cd administration | Male Wistar rats | Total brain | [75] |
| Melatonin | ↓elato ↓elat ↑elato ↑ CAT; ↑ SOD; ↑ GPX; ↑ GR | 34 mg kg−1 bw aluminum chloride orally every day for 7 days; melatonin (10 mg kg−1 bw) administered intraperitoneally every day for 7 days; the injection of melatonin was 60 min before aluminum administration | Male Wistar rats | Total brain | [109] |
| Morin | ↓ MDA; ↑ AChE; ↑ MAO; ↑ CAT; ↑ SOD; ↑ GPX; ↑ GST; ↑ GSH; ↑ vitamin C; ↑ vitamin E | 3 mg kg−1 bw cadmium chloride injected every day for 21 days; morin alone (40 mg kg−1 bw) 1 h before cadmium chloride injection for 21 days | Male Sprague-Dawley rats | Total brain | [110] |
| Morin | ↑ SOD; ↑ CAT; ↓ TBARS; ↑ GSH; ↑ GPX; | 100 mg kg−1 ammonium chloride by intraperitoneal injections thrice in a week for 8 weeks; 30 mg kg−1 morin orally by intragastric tube for 8 weeks | Male Wistar rats | Total brain | [111] |
| Taurine | ↓ MDA; ↑ CAT; ↑ SOD | Diabetes rats; 40 mg kg−1 bw streptozotocin by a single intraperitoneal injection; single dose 40 mg kg−1 bw, and after 3 days they were injected with taurine at a dose 50 mg kg−1 bw for 60 days | Male Wistar rats | Total brain | [112] |
| Taurine | ↓ ROS; ↓ MDA; ↑ GSH; ↑ CAT; ↑ SOD; ↑ GPX; ↑ AChE | Traumatic model cells; cells were treated with 100, 200, or 300 mg l−1 of taurine for 72 h | Male Wistar neonatal rats | Cortical tissues | [113] |
| Astaxanthin | ↓ MDA; ↑ GSH; ↑ SOD | 40 mg L−1 per day cadmium chloride orally for 30 days; 20 mg kg−1 per day Astaxanthin by gastric gauge for 30 days | Male Wistar rats | Total brain | [114] |
| Astaxanthin | ↑ GSH; ↑ CAT; ↓ MDA; | 2 mg kg−1 bw per week doxorubicin injected intraperitoneally (one injection per week) for 4 weeks; 25 mg kg−1 per day astaxanthin (5 days per week) orally for 4 weeks | Male albino rats | Hippocampus | [115] |
| Leonurine | ↓ MDA; ↑ GPX; ↑ SOD | Stroke rats group treated with 15, 30 or 60 mg kg−1 per day of leonurine orally once daily for 1 week | Male Sprague-Dawley rats | Total brain | [116] |
| Carvacrol | ↓ MDA; ↑ GSH; ↑ CAT; ↑ SOD; ↑ GPX; ↑ GR | Chronic restraint stress was performed using a rodent restrainer made of plexiglas that closely fit to the rats’ body (6 h per day for 21 consecutive days); 20, 30, or 40 mg kg −1 carvacrol for 21 days | Wistar rats | Total brain | [117] |
| Caffeine | ↓ LPO; ↓ ROS | 30 mg kg−1 caffeine per day for 2 weeks; 5 mg kg−1 cadmium chloride per day for 2 weeks | Male C57BL/6N mice | Hippocampus, cortex | [118] |
| Resveratrol | ↓ MDA; ↑ CAT; ↑ SOD; | 100 mg kg−1 bw aluminum chloride and 10 mg kg−1 bw sodium fluoride orally with orogastric tube for 8 weeks; 30 mg kg−1 bw resveratrol orally with orogastric tube for 8 weeks | Sprague Dawley rats | Total brain | [119] |
| Resveratrol | ↓ MDA; ↑ GSH | 375 mg kg−1 glyphosate-based herbicide in distilled water with a gastric gavage once a day for 56 days; 20 mg kg−1 resveratrol in distilled water with a gastric gavage once a day for 56 days | Male Wistar rats | Total brain | [120] |
| Protective effect | Design | Animals | Target sites | References | |
|---|---|---|---|---|---|
| Green, black, red and white tea infusion | ↑ SOD; ↑ CAT; ↑ GSH; ↑ GPX | 7 mg Cd (as cadmium chloride) and 50 mg Pb (as lead acetate) per kg of feed for 6 and 12 weeks; infusions of teas as a sole source of drink for 6 and 12 weeks | Male Wistar rats | Total brain | [35] |
| Green tea infusion | ↑ SOD; ↑ GST; ↑ GSH; ↓ NO; ↓ LPO | 0.4 % aqueous solution of lead acetate orally for 6 weeks; green tea in drinking water (15 g L−1) orally for 6 weeks | Male rats | Total brain | [130] |
| Green tea infusion | ↑ TAC; ↑ RGSH; ↑ SOD; ↓ DNA fragmentation | 100 mg of lead acetate/kg bw by gastric tube for 1 month; green tea in drinking water (5 g L−1) orally for 1 month | Albino male rats | Total rain | [129] |
| Green tea infusion | ↑ GST; ↑ RGSH; ↑ SOD; ↑ TAC; ↓ LPO | 0.4 % aqueous solution of lead acetate orally for 6 weeks; green tea in distilled water (15 g L−1) orally for 6 weeks | Rats | Total brain | [62] |
| Green tea extract | ↑ CAT; ↑ SOD; ↑ GPX; ↑ GST; ↑ total thiol; ↓ TBARS; ↓ AOPP; ↓ PC | Gasoline inhalation—2 hours daily; 1.5 % green tea extract rally as a sole source of water | Male mice CD1 strain | Total brain | [103] |
| Pomegranate peel | ↓ TBARS; ↓ NO; ↑ SOD; ↑ CAT; ↑ GPX; ↑ GR | 34 mg kg−1 bw aluminum chloride orally every day for 7 days; pomegranate peel methanolic extract (200 mg kg−1 bw) orally every day for 7 days given before aluminum chloride | Female Wistar rats | Total brain | [131] |
| Strawberry methanolic extract | ↓ LPO; ↓ NO; ↑ GSH; ↑ SOD; ↑ CAT; ↑ GPX; ↑ GR | 6.5 mg kg−1 bw per day cadmium chloride injected intraperitoneally for 5 days; 250 mg kg−1 bw per day strawberry methanolic extract orally administered 1hr before cadmium chloride injection for 5 days | Male Wistar rats | Total brain | [132] |
| Hydrophobic fractions of Thymus algeriensis | ↑ SOD; ↑ CAT; ↑ GPX; ↑ GSH; ↑ GST; ↓ LPO | 0.1 or 1 1 mmol L−1 hydrogen peroxide orally for 15 days; 180 mg hydrophobic fractions of Thymus algeriensis per kg-bw per day dissolved in normal saline orally for 15 days | Male Sprague-Dawley rats | Total brain | [133] |
| Grape seed extract and pyridoxine | ↑ SOD; ↑ CAT; ↑ GPX; ↑ GSH; ↓ MDA | 50 mg kg−1 bw per day triton injected intraperitoneally for 4 weeks; 300 mg kg−1 bw per day grape seed extract orally for 4 weeks; 12 mg kg−1 bw per day pyridoxine orally for 4 weeks | Male Sprague-Dawley rats | Total brain | [134] |
| Grape seed extract | ↑ AChE; ↓ MDA; ↑ SOD; ↑ CAT; ↑ GSH | 7.5 mg kg−1 chlorpyrifos 80% emulsion concentrate in distilled water for 21 days; 100 mg kg−1 grape seed extract orally through oral cannula for 28 days; the time interval between chlorpyrifos and grape seed extract administration was 2 h; all animals were sacrificed on day 45 | Male Wistar rats | Total brain | [135] |
| Ashwagandha extract | ↑ GSH; ↓ MDA; ↓ NO; | 100 mg−1 kg AlCl3 orally for 30 days; 200 mg−1 kg ashwagandha extract orally for 30 days; the time interval between ashwagandha and AlCl3 administration was 60 min | Male Wistar rats | Cortex, hippocampus and striatum | [136] |
| Propolis | ↓ MDA; ↓ PC; ↑ vitamin C; ↑ vitamin E; ↑ cytochrome C oxidase | 1 mg−1 kg bw lead acetate for 4 weeks; 50 mg kg−1 bw propolis orally for 4 weeks | Swiss albino rats | Total brain | [137] |
| Propolis | ↓ MDA; ↑ CAT | 0.0082 ppm cypermethrin in fresh water for 15 days; 10 ppm propolis in fresh water for 15 days | Rainbo trout Oncorhynchus mykiss | Total brain | [138] |
| Mucuna seeds extract | ↑ SOD; ↑ CAT; ↑ GSH; ↓ MDA; ↑ AChE | 2.75 mg L−1 sodium dodecyl sulphate in fresh water for 15 or 30 days; 15.5 mg kg−1 bw Mucuna extract injected intraperitoneally for 7 consecutive days | Catfish Heteropneustes fossilisand | Total brain | [139] |
| Garcinia Indica fruit extract | ↓ MDA; ↓ LPO; ↑ SOD; ↑ CAT; ↑ GSH | 75 mg kg−1 bw cyclophosphamide injected intraperitoneally 24 h before the termination of the experiment; 250 or 500 mg kg−1 bw Garcinia Indica fruit extract orally for 14 days | Male Wistar rats | Total brain | [140] |
| Chlorella vulgaris | ↑ SOD; ↑ GPX; ↑ GR; ↑ GSH; ↓ LPO | 200 mg l−1 lead acetate in drinking water for 4 weeks; 20, 50 or 100 g Chlorella vulgaris per 1 kg of food for 4 weeks | Male Sprague-Dawley rats | Total brain | [141] |
| Tomato extract, garlic extract | ↓ MDA; ↑ SOD; ↑ CAT; ↑ GSH | 6 mg kg-2 bw Cd orally for 15 days; 100 mg kg−1 bw garlic extract orally for 15 days; 50 mg kg−1 bw tomato extract orally for 15 days | Swiss albino mice | Total brain | [142] |
| Watermelon juice | ↓ MDA; ↑ GSH; ↓ CAT | 12 mL kg−1 ethanol - a single dose orally; 4 ml kg−1 watermelon juice orally for15 days before administration of ethanol | Wistar rats | Total brain | [143] |
| Parsley leaves ethanolic extract | ↓ MDA | 0.5 mL per day D-galactose injected for 20 days; 40 mg kg−1 bw ethanolic extract of parsley leaves injected for 20 days | Albino male mice | Cerebral cortex, hippocampus, cerebellum, corpora quadrigemina | [144] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Winiarska-Mieczan, A.; Baranowska-Wójcik, E.; Kwiecień, M.; Grela, E.R.; Szwajgier, D.; Kwiatkowska, K.; Kiczorowska, B. The Role of Dietary Antioxidants in the Pathogenesis of Neurodegenerative Diseases and Their Impact on Cerebral Oxidoreductive Balance. Nutrients 2020, 12, 435. https://doi.org/10.3390/nu12020435
Winiarska-Mieczan A, Baranowska-Wójcik E, Kwiecień M, Grela ER, Szwajgier D, Kwiatkowska K, Kiczorowska B. The Role of Dietary Antioxidants in the Pathogenesis of Neurodegenerative Diseases and Their Impact on Cerebral Oxidoreductive Balance. Nutrients. 2020; 12(2):435. https://doi.org/10.3390/nu12020435
Chicago/Turabian StyleWiniarska-Mieczan, Anna, Ewa Baranowska-Wójcik, Małgorzata Kwiecień, Eugeniusz R. Grela, Dominik Szwajgier, Katarzyna Kwiatkowska, and Bożena Kiczorowska. 2020. "The Role of Dietary Antioxidants in the Pathogenesis of Neurodegenerative Diseases and Their Impact on Cerebral Oxidoreductive Balance" Nutrients 12, no. 2: 435. https://doi.org/10.3390/nu12020435