Column chromatography was performed using silica gel (mesh size 40–63 µm). Thin-layer chromatography (TLC) was performed on silica gel F-254 aluminum plates and visualized using UV (254 nm/366 nm). Analytical HPLC analysis was carried out with the following systems: (system 1) Agilent 1200 HPLC (Santa Clara, CA, USA; pump G1311A, autosampler G1329A, column oven G1316A, degasser G1322A, UV detector G1315D, γ detector Gabi Star (Raytest), Luna C18 column (Phenomenex, 250 × 4.6 mm), flow rate = 1 mL/min, isocratic eluent (MeCN/0.1% TFA in H2O 70/30 (v/v)); (system 2) Agilent 1100 HPLC (Santa Clara, CA, USA; binary pump G1312A, autosampler G1313A, column oven G1316A, degasser G1322A, UV detector G1314A, γ detector Gabi Star (Raytest, Straubenhardt, Germany); column ODP-50 4B (Shodex Asahipak 50 × 4.6 mm); eluent: MeOH/PBS (10 mM, pH 7.4) gradient t0 min 30/70 - t25 min 95/5 - t27 min 95/5 - t28 min 30/70 - t40 min 30/70, flow rate = 0.6 mL/min; (system 3) waters UPLC I-Class (Milford, MA, USA; binary gradient pump BSM, autosampler FTN, column manager CM, and diode array detector PDAeλ coupled to Waters Xevo TQ-S), column Aquity UPLC® BEH C18 column (waters, 100 × 2.1 mm, 1.7 µm, 130 Å), eluent: (A): 0.1% acetic acid in MeCN/MeOH 1/1/(B): 0.1% acetic acid in H2O; flow rate 0.4 mL/min), gradient: t0 min 45/55 - t0.5 min 45/55 - t5.5 min 95/5 - t7.0 min 95/5 - t8.0 min 45/55 – t8.5 min 45/55; (system 4) column (Kinetex C-18 (Phenomenex 50 × 2.1 mm, 1.7 µm, 100 Å), Shimadzu Nexera X2 UHPLC system (Kyoto, Japan; degasser DGU-20A3R and DGU-20A5R, pump LC-30AD, autosampler SIL-30AC, column oven CTO-20AC with two column switching valves FCV-14AH, diode array detector SPD-M30A, γ detector Gabi Star (Raytest, Straubenhardt, Germany), communication bus module CBM-20A), eluent: (A): MeCN, (B): 0.1% trifluoroacetic acid in H2O; flow rate 0.5 mL/min), gradient: t0 min 25/75 - t0.3 min 25/75 - t4.0 min 75/25 - t4.5 min 95/5 - t5.5 min 95/5 - t6.0 min 25/75 – t7.5 min 25/75. The products were monitored by an UV detector at 254 nm and purity of all compounds exceeded 95% as determined by analytical HPLC analysis (system 1 or system 3), unless otherwise stated. Semi-preparative HPLC was performed using the following system: column (C-18 Jupiter Proteo (Phenomenex 250 × 21.1 mm, 4 µm, 90 Å), Shimadzu prominence modular HPLC system (Kyoto, Japan; degasser DGU-20A5R, 2× pump LC-20AR, autosampler SIL-20AC HT, column oven CTO-20AC with column switching valve, diode array detector SPD-M20A, fluorescence detector RF-20A, and fraction collector FRC-10A, communication bus module CBM-20A), isocratic eluent 0.1% TFA in MeCN/0.1% TFA in H2O 70/30, flow rate = 10 mL/min.
Low resolution mass spectra were obtained using electrospray or ASAP ionization (atmospheric solids analysis probe) using system 3. High resolution mass spectra were obtained on a Q-TOF MS using electrospray ionization: Agilent 1260 Infinity II HPLC (Santa Clara, CA, USA; pump G7111B, autosampler G7129A, column oven G7116N, UV detector G7717C, eluent MeCN/water acidified with 0.1% formic acid, bypass mode) coupled to UHD Accurate Mass Q-TOF LC MS G6538A.
Melting points were determined with a melting point apparatus (Cambridge Instruments, London, UK; GalenTM III, Testotherm testo 700; heater: Leica) and are uncorrected. Nuclear magnetic resonance spectra (NMR) were recorded on a 400 MHz (Varian, Palo Alto, CA, USA; Unity INOVA 400 MHz) spectrometer. NMR spectra were referenced to the residual solvent shifts for 1H and 13C as internal standard. DHPI and PI served as abbreviations for 1,2-dihydropyrrolo[3,2,1-hi]indole and pyrrolo[3,2,1-hi]indole, respectively.
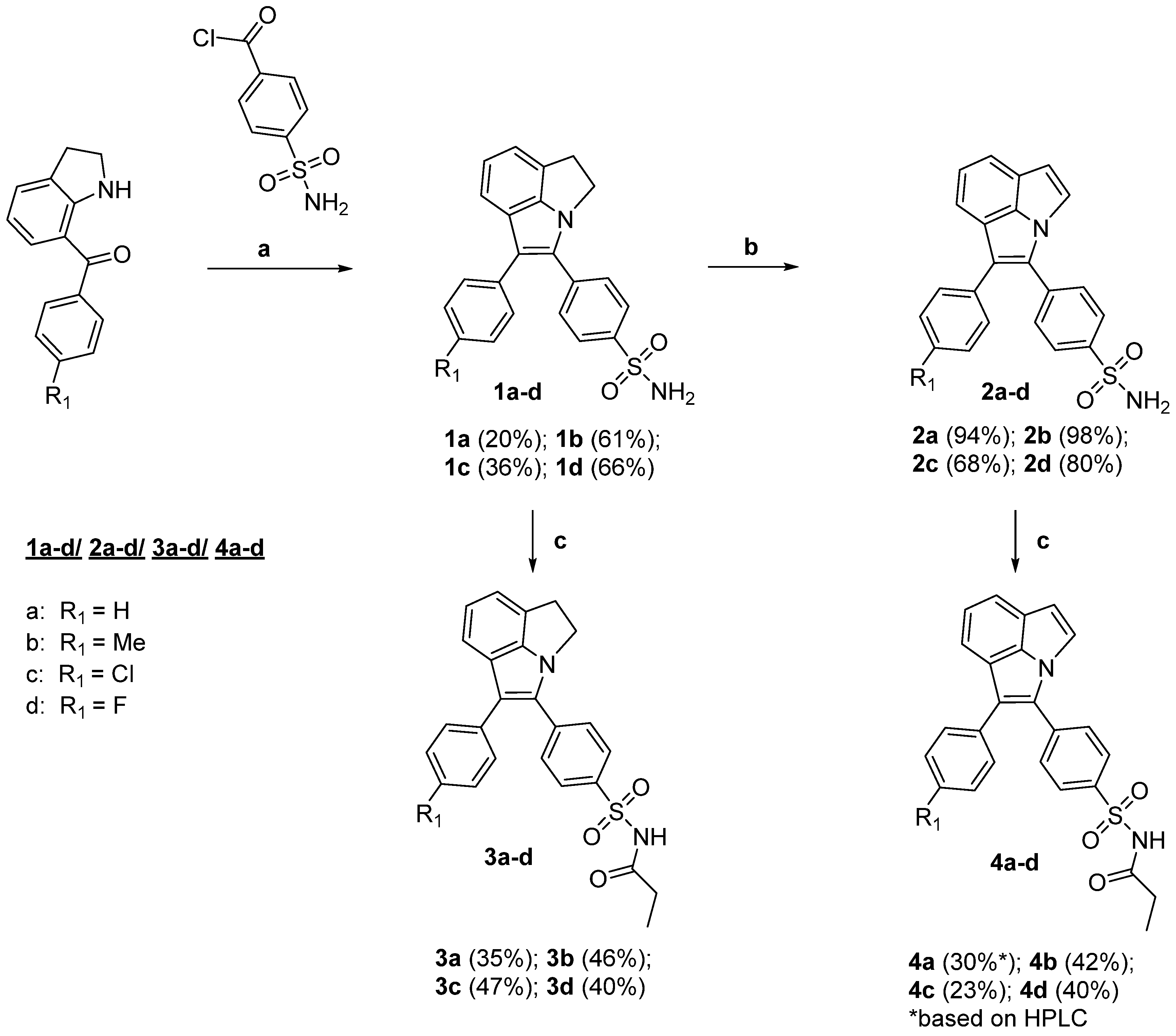
4.1. Syntheses
General Procedure A—Synthesis of 1,2-dihydropyrrolo[3,2,1-hi]indoles 1a–d:
As a starting material, 4-(sulfamoyl)benzoic acid chloride was prepared under Schlenk conditions from 4-(sulfamoyl)benzoic acid (1.504 g, 7.48 mmol, 1.0 equiv) by addition of SOCl2 (6.3 mL, 86.7 mmol, 11.5 equiv) followed by heating under reflux (60–70 °C) for 24 h. After removal of SOCl2 under reduced pressure, three times a sequence of benzene (8 mL) addition, stirring at room temperature, and removal of solvent was performed to remove traces of SOCl2. The resulting yellow solid (1.937 g, purity 85% calculated for quantitative conversion) was used without further purification for the synthesis of 1a−d.
Under Schlenk conditions, the 7-acyl-indoline (1.26 mmol, 1 equiv) was dissolved in THF (1.6 mL), followed by the addition of triethylamine (199.6 µL, 145 mg, 1.43 mmol, 1.14 equiv) and 4-(sulfamoyl)benzoic acid chloride (303 mg*, 1.38 mmol, 1.1 equiv;*the used amount of crude 4-(sulfamoyl)benzoic acid chloride was corrected for the given calculated purity) in THF (4.8 mL). The mixture was stirred at room temperature for 2 h. Then, THF (3.2 mL), zinc dust (328 mg, 5.02 mmol, 4 equiv) and TiCl4 (291.2 µL, 501 mg, 2.64 mmol, 2.1 equiv in four portions) were added and the mixture was heated to 70 °C and stirred for 2 h. After cooling, the mixture was transferred with DCM to a second flask, adsorbed on silica gel and purified by column chromatography to give the title compounds.
5-Phenyl-4-[4-(sulfamoyl)phenyl]-1,2-dihydropyrrolo[3,2,1-hi]indole (1a): Starting from 7-benzoylindoline (314 mg, 1.41 mmol, 1 equiv), 4-(sulfamoyl)benzoic acid chloride (341 mg, 1.55 mmol, 1.1 equiv), triethylamine (223.4 µL, 162 mg, 1.60 mmol, 1.14 equiv), zinc dust (368 mg, 5.63 mmol, 4 equiv) and TiCl4 (326.4 µL, 561 mg, 2.96 mmol, 2.1 equiv in four portions), the product was obtained after purification by column chromatography (1. petroleum ether/EtOAc 7/3 → 2/3; 2. petroleum ether/EtOAc 7/3 → 1/1) and sublimation in vacuo as a dark yellow solid (107 mg, 20%): mp: 220–224 °C; Rf = 0.53 (petroleum ether/EtOAc 1/2); 1H-NMR (400 MHz, (CD3)2SO): δ = 3.78 (t, 3J1,2 = 7.1 Hz, 2H, Hdhpi H1), 4.66 (t, 3J1,2 = 6.9 Hz, 2H, Hdhpi H2), 6.98 (d, 3JH,H = 6.6 Hz, 1H, Hdhpi), 7.02 (t, 3JH,H = 7.6 Hz, 3JH,H = 6.8 Hz, 1H, Hdhpi H7), 7.24 (t, 3J3,4 = 6.5 Hz, 1H, Hphenyl H4), 7.32–7.40 (m, 5H, Hdhpi/Hphenyl H2/H3/H5/H6), 7.42 (s, 2H, SO2NH2), 7.63 (d, 3J2,3 = 8.4 Hz, 1H, HSO2NH2-phenyl H2/H6), 7.82 (d, 3J2,3 = 8.5 Hz, 1H, HSO2NH2-phenyl H3/H5) ppm; 13C-NMR* (101 MHz, (CD3)2SO): δ = 32.9 (CH2), 49.8 (CH2), 116.1 (C), 116.4 (CHdhpi), 117.5 (C), 119.2 (C), 122.8 (CHdhpi), 125.5 (CHdhpi), 125.9 (CHphenyl C4), 126.1 (2CHSO2NH2-phenyl), 128.7 (2CH), 128.8 (2CH), 129.0 (2CH), 133.0 (C), 135.4 (C), 135.6 (C), 142.8 (C), 147.7 (C) ppm, *13C NMR was recorded before sublimation in vacuo and contains signals of EtOAc; MS (ASAP+): m/z (%) = 374 (100) [M]+, 375 (88) [M + H]+; HRMS (ESI/QTOF) m/z: [M + H]+ Calcd for C22H19N2O2S 375.1162, Found 375.1162; HPLC: 98.2% (tR = 7.95 min; system 1); LogD7.4 HPLC: 4.44 (tR = 24.50 ± 0.09 min).
4-[4-(Sulfamoyl)phenyl]-5-(p-tolyl)-1,2-dihydropyrrolo[3,2,1-hi]indole (1b): Starting from 7-(4-methylbenzoyl)indoline (298 mg, 1.26 mmol, 1 equiv), 4-(sulfamoyl)benzoic acid chloride (303 mg, 1.38 mmol, 1.1 equiv), triethylamine (199.6 µL, 145 mg, 1.43 mmol, 1.14 equiv), zinc dust (328 mg, 5.02 mmol, 4.0 equiv) and TiCl4 (291.2 µL, 501 mg, 2.64 mmol, 2.1 equiv in four portions), the product was obtained after purification by column chromatography (petroleum ether/EtOAc 7/3 → 2/3) as a pale yellow solid (297 mg, 61%): mp: 233–237 °C; Rf = 0.54 (petroleum ether/EtOAc 1/2); 1H-NMR (400 MHz, (CD3)2SO): δ = 2.32 (s, 3H, CH3), 3.76 (t, 3J1,2 = 6.9 Hz, 2H, Hdhpi H1), 4.64 (t, 3J1,2 = 6.9 Hz, 2H, Hdhpi H2), 6.97 (d, 3JH,H = 6.5 Hz, 1H, Hdhpi), 7.01 (t, 3JH,H = 7.5 Hz, 3JH,H = 6.8 Hz, 1H, Hdhpi H7), 7.17 (d, 3J2,3 = 8.2 Hz, 2H, Htolyl H3/H5), 7.24 (d, 3J2,3 = 8.1 Hz, 2H, Htolyl H2/H6), 7.32 (d, 3JH,H = 7.6 Hz, 1H, Hdhpi), 7.41 (s, 2H, SO2NH2), 7.62 (d, 3J2,3 = 8.4 Hz, 2H, HSO2NH2-phenyl H2/H6), 7.82 (d, 3J2,3 = 8.4 Hz, 2H, HSO2NH2-phenyl H3/H5) ppm; 13C-NMR (101 MHz, (CD3)2SO): δ = 20.7 (CH3), 32.9 (CH2), 49.8 (CH2), 116.1 (CHdhpi), 116.5 (CHdhpi), 117.6 (C), 119.2 (C), 122.7 (CHdhpi), 125.5 (C), 126.1 (2CHSO2NH2-phenyl), 128.6 (2CH), 129.0 (2CH), 129.4 (2CH), 132.6 (C), 132.8 (C), 135.0 (C), 135.6 (C), 142.7 (C), 147.7 (C) ppm; MS (ASAP+): m/z (%) = 388 (100) [M]+, 389 (51) [M + H]+; HRMS (ESI/QTOF) m/z: [M + H]+ Calcd for C23H21N2O2S 389.1318, Found 389.1320; HPLC: 99.0% (tR = 10.05 min; system 1); LogD7.4 HPLC: 4.56 (tR = 25.07 ± 0.04 min).
5-(4-Chlorophenyl)-4-[4-(sulfamoyl)phenyl]-1,2-dihydropyrrolo[3,2,1-hi]indole (1c): Starting from 7-(4-chlorobenzoyl)indoline (315 mg, 1.22 mmol, 1 equiv), 4-(sulfamoyl)benzoic acid chloride (303 mg, 1.38 mmol, 1.13 equiv), triethylamine (194 µL, 141 mg, 1.39 mmol, 1.14 equiv), zinc dust (319 mg, 4.74 mmol, 3.9 equiv) and TiCl4 (283.2 µL, 487 mg, 2.57 mmol, 2.1 equiv in four portions), the product was obtained after purification by column chromatography (petroleum ether/EtOAc 7/3 → 3/2) as a pale yellow solid (182 mg, 36%): mp: 259–265 °C (degradation starting at 265 °C); Rf = 0.53 (petroleum ether/EtOAc 1/2; 1H-NMR (400 MHz, (CD3)2SO): δ = 3.77 (t, 3J1,2 = 6.7 Hz, 2H, Hdhpi H1), 4.65 (t, 3J1,2 = 6.8 Hz, 2H, Hdhpi H2), 6.99 (d, 3JH,H = 6.7 Hz, 1H, Hdhpi), 7.04 (t, 3JH,H = 7.7 Hz, 3JH,H = 6.8 Hz, 1H, Hdhpi H7), 7.31–7.38 (m, 3H, 1Hdhpi/2Hchlorphenyl H3/H5), 7.42 (d, 3J2,3 = 8.4 Hz, 4H, NH2/2Hchlorphenyl H2/H6), 7.64 (d, 3J2,3 = 8.3 Hz, 2H, HSO2NH2-phenyl H2/H6), 7.85 (d, 3J2,3 = 8.3 Hz, 2H, HSO2NH2-phenyl H3/H5) ppm; 13C-NMR (101 MHz, (CD3)2SO): δ = 32.9 (CH2), 49.7 (CH2), 116.0 (C), 116.3 (CHdhpi/C)*, 119.0 (C), 123.0 (CHdhpi), 125.6 (C), 126.2 (2CHSO2NH2-phenyl), 128.8 (2CH), 129.1 (2CH), 130.3 (2CH), 130.4 (CHdhpi), 133.3 (C), 134.5 (C), 135.1 (C), 143.0 (C), 147.6 (C) ppm, *two carbon species with identical chemical shift; MS (ASAP+): m/z (%) = 101 (100), 408 (73) [M]+, 409 (44) [M + H]+; HRMS (ESI/QTOF) m/z: [M + H, 35Cl]+ Calcd for C22H18ClN2O2S 409.0772, Found 409.0773; HPLC: 93.4% (tR = 11.23 min; system 1); LogD7.4 HPLC: 4.90 (tR = 26.76 ± 0.06 min).
5-(4-Fluorophenyl)-4-[4-(sulfamoyl)phenyl]-1,2-dihydropyrrolo[3,2,1-hi]indole (1d): Starting from 7-(4-fluorobenzoyl)indoline (304 mg, 1.26 mmol, 1 equiv), 4-(sulfamoyl)benzoic acid chloride (304 mg, 1.38 mmol, 1.1 equiv), triethylamine (200.2 µL, 145 mg, 1.44 mmol, 1.14 equiv), zinc dust (330 mg, 5.05 mmol, 4 equiv) and TiCl4 (292 µL, 502 mg, 2.65 mmol, 2.1 equiv in four portions), the product was obtained after purification by column chromatography (petroleum ether/EtOAc 7/3 → 1/1) as a pale yellow solid (328 mg, 66%): mp: 250–252 °C; Rf = 0.54 (petroleum ether/EtOAc 1/2); 1H-NMR (400 MHz, (CD3)2SO): δ = 3.77 (t, 3J1,2 = 6.9 Hz, 2H, Hdhpi H1), 4.65 (t, 3J1,2 = 6.9 Hz, 2H, Hdhpi H2), 6.98 (d, 3JH,H = 6.7 Hz, 1H, Hdhpi), 7.03 (t, 3JH,H = 7.6 Hz, 3JH,H = 6.8 Hz, 1H, Hdhpi H7), 7.21 (t, 3J2,3 = 8.9 Hz, 3J3,F = 8.9 Hz, 2H, HF-phenyl H3/H5), 7.33 (d, 3JH,H = 7.7 Hz, 1H, Hdhpi), 7.36 (dd, 3J2,3 = 8.8 Hz, 4J2,F = 5.6 Hz, 2H, HF-phenyl H2/H6), 7.42 (s, 2H, SO2NH2), 7.62 (d, 3J2,3 = 8.5 Hz, 2H, HSO2NH2-phenyl H3/H5), 7.84 (d, 3J2,3 = 8.5 Hz, 2H, HSO2NH2-phenyl H2/H6) ppm; 13C-NMR (101 MHz, (CD3)2SO): δ = 32.9 (CH2), 49.8 (CH2), 115.7 (d, 2J3,F = 21 Hz, CHF-phenyl C3/C5), 116.2 (CHdhpi), 116.3 (CHdhpi), 116.4 (C), 119.1 (C), 122.9 (CHdhpi), 125.6 (C), 126.2 (2CHSO2NH2-phenyl), 129.0 (2CHSO2NH2-phenyl), 130.5 (d, 3J2,F = 8 Hz, CHF-phenyl C2/C6), 132.0 (d, 4J1,F = 3 Hz, CF-phenyl C1), 133.1 (C), 135.2 (C), 142.9 (C), 147.6 (C), 160.6 (d, 1J4,F = 243 Hz, CF-phenyl C4) ppm; 19F-NMR (376 MHz, (CD3)2SO): δ = −116.6 ppm; MS (ASAP+): m/z (%) = 392 (49) [M]+, 393 (100) [M + H]+; HRMS (ESI/QTOF) m/z: [M + H]+ Calcd for C22H18FN2O2S 393.1068, Found 393.1066; HPLC: 98.9% (tR = 8.29 min; system 1); LogD7.4 HPLC: 4.53 (tR = 24.92 ± 0.01 min).
General Procedure B—Synthesis of pyrrolo[3,2,1-hi]indoles 2a–d:
Under nitrogen atmosphere, DDQ (142 mg, 0.63 mmol, 3.5 equiv) in benzene (8.5 mL) was added to the 1,2-dihydropyrrolo[3,2,1-hi]indole 1a–d (0.18 mmol, 1.0 equiv) in benzene (2 mL) in a Schlenk flask. The mixture was heated to 100°C and stirred at this temperature for 6 h. After cooling, the mixture was transferred with EtOAc (20 mL) into a separation funnel. The organic phase was washed with saturated sodium thiosulfate (20 mL), saturated sodium bicarbonate (20 mL), and brine (10 mL). The aqueous phase was combined and extracted with EtOAc (2 × 20 mL). After drying of the combined organic phase over sodium sulfate, the crude product was adsorbed on silica gel and further purified by column chromatography as given below to give the title compounds 2a–d.
1-Phenyl-2-[4-(sulfamoyl)phenyl]pyrrolo[3,2,1-hi]indole (2a): Starting from 1a (64.0 mg, 0.17 mmol, 1 equiv) and DDQ (136 mg, 0.6 mmol, 3.52 equiv), the product was obtained after purification by column chromatography (petroleum ether/EtOAc 7/3) as a yellow solid (59.6 mg, 94%): mp: 218–220°C; Rf = 0.44 (petroleum ether/EtOAc 1/1); 1H-NMR (400 MHz, (CD3)2)SO): δ = 7.00 (d, 3J4,5 = 3.1 Hz, 1H, Hpi H5), 7.37 (t, 3J3,4 = 7.1 Hz, 1H, Hphenyl H4), 7.46 (t, 3JH,H = 7.4 Hz, 3JH,H = 7.8 Hz, 2H, Hphenyl H3/H5), 7.48–7.56 (m, 5H, SO2NH2/Hpi H7/Hphenyl H2/H6), 7.73–7.79 (m, 3H, 1Hpi/HSO2NH2-phenyl H2/H6), 7.81 (d, 3JH,H = 7.3 Hz, 1H, Hpi), 7.90 (d, 3J2,3 = 8.4 Hz, 2H, HSO2NH2-phenyl H3/H5), 7.94 (d, 3J4,5 = 3.1 Hz, 1H, Hpi H4) ppm; 13C-NMR (101 MHz, (CD3)2SO): δ = 111.0 (C), 119.7 (CHpi), 121.1 (CHpi), 122.0 (C), 122.1 (CHpi), 123.3 (C), 124.8 (CHpi), 125.9 (CHpi), 126.4 (2CHSO2NH2-phenyl), 127.2 (CHphenyl C4), 129.1 (2CH), 129.2 (2CH), 129.7 (2CH), 133.0 (C), 133.9 (C), 133.9 (C), 136.4 (C), 143.7 (C) ppm; MS (ASAP+): m/z (%) = 372 (100) [M]+; MS (ASAP+): m/z (%) = 374 (100) [M]+, 375 (88) [M + H]+; HRMS (ESI/QTOF) m/z: [M + H]+ Calcd for C22H17N2O2S 373.1005, Found 373.1006; HPLC: 99.3% (tR = 8.77 min; system 1); LogD7.4 HPLC: 4.59 (tR = 25.22 ± 0.02 min).
2-[4-(Sulfamoyl)phenyl]-1-(p-tolyl)pyrrolo[3,2,1-hi]indole (2b): Starting from 1b (100 mg, 0.26 mmol, 1 equiv) and DDQ (204 mg, 0.90 mmol, 3.46 equiv), the product was obtained after purification by column chromatography (petroleum ether/EtOAc 7/3) as a beige solid (97 mg, 98%): mp: 233–235°C; Rf = 0.47 (petroleum ether/EtOAc 1/1); 1H-NMR (400 MHz, (CD3)2SO): δ = 2.36 (s, 3H, CH3 tolyl), 6.98 (d, 3J4,5 = 3.1 Hz, 1H, Hpi H5), 7.26 (d, 3J2,3 = 8.3 Hz, 2H, Htolyl H3/H5), 7.39 (d, 3J2,3 = 8.1 Hz, 2H, Htolyl H2/H6), 7.46–7.54 (m, 3JH,H = 7.3 Hz, 3H, SO2NH2/Hpi H7), 7.72–7.78 (m, 3J2,3 = 8.4 Hz, 3H, Hpi/HSO2NH2-phenyl H2/H6), 7.80 (d, 3JH,H = 7.3 Hz, 1H, Hpi), 7.90 (d, 3J2,3 = 8.3 Hz, 2H, H SO2NH2-phenyl H3/H5), 7.93 (d, 3J4,5 = 3.1 Hz, 1H, Hpi H4) ppm; 13C-NMR (101 MHz, (CD3)2SO): δ = 20.8 (CH3), 110.7 (CHpi), 119.6 (CHpi), 121.1 (CHpi), 122.1 (C/C)*, 123.3 (C), 124.6 (CHpi), 125.8 (CHpi), 126.4 (2CHSO2NH2-phenyl), 129.0 (2CH), 129.6 (2CH), 129.7 (2CH), 130.9 (C), 132.7 (C), 134.0 (C), 136.4 (C), 136.5 (C), 143.6 (C) ppm, *two carbon species with identical chemical shift; MS (ASAP+): m/z (%) = 101 (100), 386 (91) [M]+, 387 (31) [M + H]+; HRMS (ESI/QTOF) m/z: [M + H]+ Calcd for C23H19N2O2S 387.1162, Found 387.1163; HPLC: 99.1% (tR = 11.47 min; system 1); LogD7.4 HPLC: 4.73 (tR = 25.91 ± 0.01 min).
1-(4-Chlorophenyl)-2-[4-(sulfamoyl)phenyl]pyrrolo[3,2,1-hi]indole (2c): Starting from 1c (91 mg, 0.22 mmol, 1 equiv) and DDQ (177 mg, 0.78 mmol, 3.55 equiv), the product was obtained after purification by column chromatography (petroleum ether/EtOAc 4:1 → 7/3) as a pale yellow solid (62 mg, 68%): mp: 270-273°C; Rf = 0.48 (petroleum ether/EtOAc 1/1); 1H-NMR (400 MHz, (CD3)2SO): δ = 7.00 (d, 3J4,5 = 2.7 Hz, 1H, Hpi H5), 7.47–7.56 (m, 7H, SO2NH2/Hpi H7/HCl-phenyl H2/H3/H5/H6), 7.74–7.80 (m, 3H, Hpi/HSO2NH2-phenyl H2/H6), 7.82 (d, 3JH,H = 7.3 Hz, 1H, Hpi), 7.89–7.96 (m, 3H, Hpi H4/HSO2NH2-phenyl H3/H5) ppm; 13C-NMR (101 MHz, (CD3)2SO): δ = 111.1 (CHpi), 119.6 (CHpi), 121.3 (CHpi), 121.7 (C), 121.9 (C), 122.1 (C), 124.8 (CHpi), 125.7 (CHpi), 126.5 (2CHSO2NH2-phenyl), 129.1 (2CH), 129.7 (2CH), 130.9 (2CH), 131.9 (C), 132.9 (C), 133.3 (C), 133.6 (C), 136.3 (C), 143.8 (C) ppm; MS (ASAP+): m/z (%) = 101 (100), 406 (49) [M, 35Cl]+, 409 (22) [M, 37Cl]+; HRMS (ESI/QTOF) m/z: [M + H, 35Cl]+ Calcd for C22H16ClN2O2S 407.0616, Found 407.0613; HPLC: 97.1% (tR = 7.07 min; acetonitrile/0.1% TFA in water 80:20, system 1); LogD7.4 HPLC: 4.89 (tR = 26.71 ± 0.01 min).
1-(4-Fluorophenyl)-2-[4-(sulfamoyl)phenyl]pyrrolo[3,2,1-hi]indole (2d): Starting from 1d (100 mg, 0.25 mmol, 1 equiv) and DDQ (203 mg, 0.89 mmol, 3.56 equiv), the product was obtained after purification by column chromatography (petroleum ether/EtOAc 7/3 → 3/2 → 1/1) as a pale yellow solid (79 mg, 80%): mp: 258-259 °C; Rf = 0.49 (petroleum ether/EtOAc 1/1); 1H-NMR (400 MHz, (CD3)2SO): δ = 7.00 (d, 3J4,5 = 3.1 Hz, 1H, Hpi H5), 7.30 (t, 3J2,3 = 8.9 Hz, 3J3,F = 8.9 Hz, 2H, HF-phenyl H3/H5), 7.47–7.58 (m, 4J2,F = 5.4 Hz, 5H, 1Hpi H7/SO2NH2/HF-phenyl H2/H6), 7.73–7.78 (m, 3JH,H = 7.2 Hz, 3J2,3 = 8.4 Hz, 3H, Hpi/HSO2NH2-phenyl H2/H6), 7.81 (d, 3JH,H = 7.3 Hz, 1H, Hpi), 7.91 (d, 3J2,3 = 8.4 Hz, 2H, HSO2NH2-phenyl H3/H5), 7.94 (d, 3J4,5 = 3.2 Hz, 1H, Hpi H4) ppm; 13C-NMR (101 MHz, (CD3)2SO): δ = 111.0 (CHpi), 116.0 (d, 2J3,F = 22 Hz, CHF-phenyl C3/C5), 119.5 (CHpi), 121.2 (CHpi), 121.9 (C), 122.1 (C), 122.2 (C), 124.7 (CHpi), 125.9 (CHpi), 126.4 (2CHSO2NH2-phenyl), 129.6 (2CHSO2NH2-phenyl), 130.3 (d, 4J1,F = 3 Hz, CF-phenyl C1), 131.2 (d, 3J2,F = 8 Hz, CHF-phenyl C2/C6), 133.0 (C), 133.7 (C), 136.3 (C), 143.7 (C), 161.4 (d, 1J4,F = 244 Hz, CF-phenyl C4) ppm; 19F-NMR (376 MHz, (CD3)2SO): δ = -110.1 ppm; MS (ASAP+): m/z (%) = 101 (100), 390 (94) [M]+, 391 (34) [M + H]+; HRMS (ESI/QTOF) m/z: [M + H]+ Calcd for C22H16FN2O2S 391.0911, Found 391.0906; HPLC: 98.8% (tR = 9.36 min; system 1); LogD7.4 HPLC: 4.64 (tR = 25.49 ± 0.05 min).
General Procedure C—Synthesis of N-propionamide-substituted 1,2-dihydropyrrolo[3,2,1-hi]indoles 3a–d and pyrrolo[3,2,1-hi]indoles 4a–d.
The (dihydro)pyrrolo[3,2,1-hi]indole 1a–d or 2a–d (25 µmol, 1.0 equiv) and 4-dimethylaminopyridine (DMAP, 6.17 mg, 50 µmol, 2 equiv) were dissolved in anhydrous THF (760 µL) and anhydrous DCM (760 µL). Then, propionyl chloride (2.86 µL, 3.06 mg, 33.1 µmol, 1.31 equiv) was added and the solution was stirred at room temperature for 20 min. Reaction control at this time point by HPLC indicated complete consumption of the starting material. Afterwards, the solvent was evaporated at room temperature in a stream of nitrogen and the crude product was redissolved in MeCN/0.1% TFA in water and purified by semi-preparative HPLC (0.1% TFA in MeCN/0.1% TFA in water 70/30 (v/v)) to give the title compounds 3a–d and 4a–d.
N-{[4-(5-phenyl-1,2-dihydropyrrolo[3,2,1-hi]indol-4-yl)phenyl]sulfonyl}propionamide (3a): Starting from 1a (9.46 mg, 25.26 µmol, 1 equiv), DMAP (6.17 mg, 50.53 µmol, 2.0 equiv), and propionyl chloride (2.89 µL, 33.2 µmol, 1.3 equiv) and following general procedure C, the product was obtained after semi-preparative HPLC (tR = 15 min) as a yellow solid (3.79 mg, 35%): mp: 212-214 °C, unstable crystal modification melted at 108-111 °C (from lyophilisation); Rf = 0.29 (n-hexane/EtOAc 1/1); 1H-NMR (400 MHz, Chloroform-d): δ = 1.12 (t, 3JH,H = 7.4 Hz, 3H, CH2CH3), 2.33 (q, 3JH,H = 7.4 Hz, 2H, CH2CH3), 3.84 (t, 3J1,2 = 7.0 Hz, 2H, Hdhpi H1), 4.61 (t, 3J1,2 = 7.2 Hz, 2H, Hdhpi H2), 7.01 (dd, 3JH,H = 6.7 Hz, 4JH,H = 0.8 Hz, 1H, Hdhpi), 7.08 (dd, 3JH,H = 7.9, 3JH,H = 6.8 Hz, 1H, Hdhpi H7), 7.28 (t, 4JH,H = 1.5 Hz, 0.3H, part of Hphenyl H4)*, 7.35 (t, 3JH,H = 7.6 Hz, 2H, Hphenyl H3/H5), 7.41 (dd, 3J3,4 = 8.3 H, 4J2,4 = 1.4 Hz, 2H, Hphenyl H2/H6), 7.44 (dd, 3JH,H = 7.9 Hz, 4JH,H = 0.7 Hz, 1H, Hdhpi), 7.57 (d, 3JH,H = 8.8 Hz, 2H, HSO2-phenyl H2/H6), 7.94 (s, 1H, NH), 7.99 (d, 3JH,H = 8.8 Hz, 2H, HSO2-phenyl H3/H5) ppm, *part of Hphenyl H4 signal which overlaps with residual solvent signal of CDCl3; 13C-NMR (101 MHz, Chloroform-d): δ = 8.4 (CH2CH3), 29.8 (CH2CH3), 33.7 (CH2), 50.7 (CH2), 116.8 (CHdhpi), 117.6 (CHdhpi), 120.3 (C), 120.5 (C), 123.2 (CHdhpi), 125.1 (C), 126.5 (CHphenyl H4), 128.8 (2CHSO2-phenyl), 128.9 (2CHphenyl), 129.2 (2CHSO2-phenyl), 129.5 (2CHphenyl), 133.0 (C), 135.7 (C), 136.7 (C), 138.9 (C), 148.9 (C), 171.1 (CONH) ppm; MS (ASAP+): m/z (%) = 374 (100) [M]+, 375 (88) [M + H]+; MS (ESI+): m/z (%) = 296.3 (11) [M + H-SO2NHCOC2H5]+, 431.3 (100) [M + H]+, 533.9 (15); HRMS (ESI/QTOF) m/z: [M + H]+ Calcd for C25H23N2O3S 431.1424, Found 431.1416; HPLC: 98.8% (tR = 4.56 min; system 3); LogD7.4 HPLC: 2.05 (tR = 13.38 ± 0.08 min).
N-({4-[5-(p-tolyl)-1,2-dihydropyrrolo[3,2,1-hi]indol-4-yl]phenyl}sulfonyl)propionamide (3b): Starting from 1b (9.52 mg, 24.51 µmol, 1 equiv), DMAP (5.98 mg, 49.01 µmol, 2.0 equiv), and propionyl chloride (2.80 µL, 32.1 µmol, 1.3 equiv) and following general procedure C, the product was obtained after semi-preparative HPLC (tR = 19 min) as a yellow solid (4.96 mg, 46%): mp: 116-117 °C (from lyophilisation); Rf = 0.28 (n-hexane/EtOAc 1/1); 1H-NMR (400 MHz, Chloroform-d): δ = 1.12 (t, 3JH,H = 7.4 Hz, 3H, CH2CH3), 2.34 (q, 3JH,H = 7.4 Hz, 2H, CH2CH3), 2.38 (s, 3H, CH3), 3.83 (t, 3J1,2 = 7.0 Hz, 2H, Hdhpi H1), 4.59 (dd, 3J1,2 = 7.5, 6.5 Hz, 2H, Hdhpi H2), 7.00 (d, 3JH,H = 7.1 Hz, 1H, Hdhpi), 7.07 (dd, 3JH,H = 7.9, 6.7 Hz, 1H, Hdhpi H7), 7.16 (d, 3J2,3 = 7.8 Hz, 2H, Htolyl H3/H5), 7.30 (d, 3J2,3 = 8.1 Hz, 2H, Htolyl H2/H6), 7.43 (dd, 3JH,H = 7.9, 0.7 Hz, 1H, Hdhpi), 7.58 (d, 3J2,3 = 8.7 Hz, 2H, HSO2-phenyl H2/H6), 7.99 (d, 3J2,3 = 8.6 Hz, 2H, HSO2-phenyl H3/H5), 8.01 (s, 1H, NH) ppm; 13C-NMR (101 MHz, Chloroform-d): δ = 8.4 (CH2CH3), 21.4 (CH3), 29.8 (CH2CH3), 33.7 (CH2), 50.7 (CH2), 116.8 (CHdhpi), 117.6 (CHdhpi), 120.4 (C), 120.6 (C), 123.0 (CHdhpi), 125.0 (C), 128.7 (2CHSO2-phenyl), 129.1 (2CHSO2-phenyl), 129.4 (2CHtolyl), 129.7 (2CHtolyl), 132.7 (C), 132.8 (C), 136.2 (C), 136.6 (C), 139.1 (C), 148.9 (C), 171.2 (CONH) ppm; MS (ESI+): m/z (%) = 309.3 (9) [M + H-SO2NHCOC2H5]+, 445.5 (100) [M + H]+; HRMS (ESI/QTOF) m/z: [M + H]+ Calcd for C26H25N2O3S 445.1581, Found 445.1576; HPLC: 99.5% (tR = 4.56 min; system 3); LogD7.4 HPLC: 2.31 (tR = 13.92 ± 0.08 min).
N-({4-[5-(4-chlorophenyl)-1,2-dihydropyrrolo[3,2,1-hi]indol-4-yl]phenyl}sulfonyl)propionamide (3c): Starting from 1c (9.32 mg, 22.79 µmol, 1 equiv), DMAP (5.57 mg, 45.59 µmol, 2.0 equiv), and propionyl chloride (2.61 µL, 29.88 µmol, 1.3 equiv) and following general procedure C, the product was obtained after semi-preparative HPLC (tR = 20 min) as a yellow solid (5.02 mg, 47%): mp: 232–235 °C, unstable crystal modification melted at 120-122°C (from lyophilisation); Rf = 0.28 (n-hexane/EtOAc 1/1); 1H-NMR (400 MHz, Chloroform-d): δ = 1.13 (t, 3JH,H = 7.4 Hz, 3H, CH2CH3), 2.34 (q, 3JH,H = 7.4 Hz, 2H, CH2CH3), 3.84 (t, 3J1,2 = 7.0 Hz, 2H, Hdhpi H1), 4.60 (t, 3J1,2 = 7.0 Hz, 2H, Hdhpi H2), 7.02 (d, 3JH,H = 7.0 Hz, 1H, Hdhpi), 7.09 (dd, 3JH,H = 7.9, 6.8 Hz, 1H, Hdhpi H7), 7.27–7.37 (m, 4H, HCl-phenyl), 7.40 (dd, 3JH,H = 7.9 Hz, 4JH,H = 0.7 Hz, 1H, Hdhpi), 7.56 (d, 3J2,3 = 8.8 Hz, 2H, HSO2-phenyl H2/H6), 7.98–8.05 (m, 3H, NH/2HSO2-phenyl H3/H5) ppm; 13C-NMR (101 MHz, Chloroform-d): δ = 8.4 (CH2CH3), 29.8 (CH2CH3), 33.7 (CH2), 50.7 (CH2), 117.0 (CHdhpi), 117.3 (CHdhpi), 119.1 (C), 120.1 (C), 123.4 (CHdhpi), 125.1 (C), 128.9 (2CHSO2-phenyl), 129.2 (2CH), 129.2 (2CH), 130.7 (2CHCl-phenyl), 132.2 (C), 133.1 (C), 134.3 (C), 137.1 (C), 138.6 (C), 148.8 (C), 171.2 (CONH) ppm; MS (ESI+): m/z (%) = 329.5 (26) [M + H-SO2NHCOC2H5, 35Cl]+, 465.2 (100) [M + H, 35Cl]+; HRMS (ESI/QTOF) m/z: [M + H, 35Cl]+ Calcd for C25H22ClN2O3S 465.1034, Found 465.1029; HPLC: 99.2% (tR = 5.04 min; system 3); LogD7.4 HPLC: 2.49 (tR = 14.81 ± 0.00 min).
N-({4-[5-(4-fluorophenyl)-1,2-dihydropyrrolo[3,2,1-hi]indol-4-yl]phenyl}sulfonyl)propionamide (3d): Starting from 1d (10.19 mg, 25.97 µmol, 1 equiv), DMAP (6.34 mg, 51.93 µmol, 2.0 equiv), and propionyl chloride (2.97 µL, 34.04 µmol, 1.3 equiv) and following general procedure C, the product was obtained after semi-preparative HPLC (tR = 15 min) as a pale yellow solid (4.70 mg, 40%): mp: 119-126 °C (from lyophilisation); Rf = 0.26 (n-hexane/EtOAc 1/1); 1H-NMR (400 MHz, Chloroform-d): δ = 1.13 (t, 3JH,H = 7.4 Hz, 3H, CH2CH3), 2.33 (q, 3JH,H = 7.4 Hz, 2H, CH2CH3), 3.84 (t, 3J1,2 = 7.0 Hz, 2H, Hdhpi H1), 4.60 (t, 3J1,2 = 7.0 Hz, 2H, Hdhpi H2), 7.00–7.11 (m, 4H, 2Hdhpi/HF-phenyl H3/H5), 7.35 (dd, 3J2,3 = 8.9 Hz, 4J2,F = 5.4 Hz, 2H, H F-phenyl H2/H6), 7.39 (d, 3JH,H = 8.2 Hz, 1H, Hdhpi), 7.55 (d, 3J2,3 = 8.8 Hz, 2H, HSO2-phenyl H2/H6), 7.94 (s, 1H, NH), 8.00 (d, 3J2,3 = 8.8 Hz, 2H, HSO2-phenyl H3/H5) ppm; 13C-NMR (101 MHz, Chloroform-d): δ = 8.4 (CH2CH3), 29.8 (CH2CH3), 33.7 (CH2), 50.7 (CH2), 115.9 (d, 2J3,F = 22 Hz, CHF-phenyl C3/C5), 116.9 (CHdhpi), 117.3 (CHdhpi), 119.4 (C), 120.3 (C), 123.3 (CHdhpi), 125.1 (C), 128.9 (2CHSO2-phenyl), 129.1 (2CHSO2-phenyl), 131.0 (d, 3J2,F = 8 Hz, CHF-phenyl C2/C6), 131.7 (d, 4J1,F = 3 Hz, CF-phenyl C1), 133.0 (C), 136.9 (C), 138.7 (C), 148.8 (C), 171.1 (CONH) ppm, signal of 1 quaternary carbon not resolved; 19F-NMR (376 MHz, Chloroform-d): δ = −116.0 ppm; MS (ESI+): m/z (%) = 313.3 (26) [M + H-SO2NHCOC2H]+, 449.1 (100) [M + H]+; HRMS (ESI/QTOF) m/z: [M + H]+ Calcd for C25H22FN2O3S 449.1330, Found 449.1329; HPLC: 99.6% (tR = 4.64 min; system 3); LogD7.4 HPLC: 2.20 (tR = 13.38 ± 0.08 min).
N-{[4-(1-phenylpyrrolo[3,2,1-hi]indol-2-yl)phenyl]sulfonyl}propionamide (4a): Starting from 2a (0.86 mg, 2.31 µmol, 1 equiv), DMAP (0.56 mg, 4.62 µmol, 2.0 equiv), and propionyl chloride (0.26 µL, 3.02 µmol, 1.3 equiv) and following general procedure C, the conversion as indicated by UPLC was incomplete after the indicated reaction time. After portionwise addition of further propionyl chloride (4× 0.26 µL, 3.02 µmol, 1.3 equiv, then 1× 40 µL, 464 µmol, 200 equiv) and DMAP (0.56 mg, 4.62 µmol, 2.0 equiv) and stirring at room temperature for 20 min intervals, the reaction was stopped and the product was obtained after semi-preparative HPLC (tR = 17 min) as a solid (30% conversion based on HPLC system 4): Rf = 0.34 (n-hexane/EtOAc 1/1); MS (ESI+): m/z (%) = 293.2 (100) [M + H-SO2NHCOC2H]+, 429.2 (77) [M + H]+, 451.2 (58) [M + Na]+; HRMS (ESI/QTOF) m/z: [M + H]+ Calcd for C25H21N2O3S 429.1268, Found 429.1262; HPLC: 100% (tR = 4.76 min; system 3); LogD7.4 HPLC: 2.24 (tR = 13.56 ± 0.05 min).
N-({4-[1-(p-tolyl)pyrrolo[3,2,1-hi]indol-2-yl]phenyl}sulfonyl)propionamide (4b): Starting from 2b (11.27 mg, 29.16 µmol, 1 equiv), DMAP (7.12 mg, 58.32 µmol, 2.0 equiv), and propionyl chloride (3.33 µL, 38.20 µmol, 1.3 equiv) and following general procedure C, the product was obtained after semi-preparative HPLC (tR = 20 min) as a yellow solid (5.44 mg, 42%): mp: 110-115 °C (from lyophilisation); Rf = 0.35 (n-hexane/EtOAc 1/1); 1H-NMR (400 MHz, (Chloroform-d): δ = 1.13 (t, 3JH,H = 7.4 Hz, 3H), 2.35 (q, 3JH,H = 7.4 Hz, 2H, CH2CH3), 2.41 (s, 3H, CH3), 6.89 (d, 3J4,5 = 3.1 Hz, 1H, Hpi), 7.22 (d, 3J2,3 = 7.8 Hz, 2H, Htolyl), 7.39 (d, 3J2,3 = 8.0 Hz, 2H, Htolyl), 7.51 (t, 3JH,H = 7.4 Hz, 1H, Hpi H7), 7.57 (d, 3J4,5 = 3.1 Hz, 1H, Hpi), 7.73 (d, 3J2,3 = 8.8 Hz, 2H, HSO2-phenyl)*, 7.73–7.80 (m, 2H, 2Hpi), 8.04–8.11 (m, 3H, NH/HSO2-phenyl)* ppm, *integral 1.5 H, two further aromatic signals at 7.59 (d, J = 8.2 Hz, ‘0.6H’) and 7.97 (d, J = 8.5 Hz, ‘0.6H’) were detected that account for the 2-phenyl ring of the deprotonated species; MS (ESI+): m/z (%) = 307.2 (98) [M + H-SO2NHCOC2H]+, 443.2 (100) [M + H]+; HRMS (ESI/QTOF) m/z: [M + H]+ Calcd for C26H22N2O3S 443.1424, Found 443.1418; HPLC: 99.7% (tR = 5.12 min; system 3); LogD7.4 HPLC: 2.48 (tR = 14.75 ± 0.01 min).
N-({4-[1-(4-chlorophenyl)pyrrolo[3,2,1-hi]indol-2-yl]phenyl}sulfonyl)propionamide (4c): Starting from 2c (1.01 mg, 2.48 µmol, 1 equiv), DMAP (0.61 mg, 4.96 µmol, 2.0 equiv), and propionyl chloride (0.28 µL, 3.25 µmol, 1.3 equiv) and following general procedure C, the conversion as indicated by UPLC was incomplete after the indicated reaction time. After addition of further propionyl chloride (1 × 40 µL, 464 µmol, 187 equiv) and DMAP (0.56 mg, 4.62 µmol, 2.0 equiv) and stirring at room temperature for 20 min, the reaction was stopped and the product was obtained after semi-preparative HPLC (tR = 22 min) as a yellow solid (0.26 mg, 23%): Rf = 0.34 (n-hexane/EtOAc 1/1); MS (ESI+): m/z (%) = 327.2 (100) [M + H-SO2NHCOC2H5, 35Cl]+, 409.1 (91), 463.1 (78) [M + H, 35Cl]+; 485.1 (93) [M + Na, 35Cl]+; HRMS (ESI/QTOF) m/z: [M + H, 35Cl]+ Calcd for C25H20ClN2O3S 463.0878, Found 463.0875; HPLC: 100% (tR = 5.17 min; system 3); LogD7.4 HPLC: 2.60 (tR = 15.37 min).
N-({4-[1-(4-fluorophenyl)pyrrolo[3,2,1-hi]indol-2-yl]phenyl}sulfonyl)propionamide (4d): Starting from 2d (11.62 mg, 29.76 µmol, 1 equiv), DMAP (7.27 mg, 59.52 µmol, 2.0 equiv), and propionyl chloride (3.40 µL, 38.99 µmol, 1.3 equiv) and following general procedure C, the product was obtained after semi-preparative HPLC (tR = 17 min) as a yellow solid (5.32 mg, 40%): mp: 202–206 °C; Rf = 0.35 (n-hexane/EtOAc 1/1); 1H-NMR (400 MHz, Chloroform-d): δ = 1.14 (t, 3JH,H = 7.4 Hz, 3H, CH2CH3), 2.34 (q, 3JH,H = 7.3 Hz, 2H, CH2CH3), 6.91 (d, 3J4,5 = 3.1 Hz, 1H, Hpi), 7.11 (t, 3J2,3 = 3J3,F = 8.8 Hz, 2H, HF-phenyl H3/H5), 7.46 (dd, 3J2,3 = 8.8, 4J2,F = 5.4 Hz, 2H, HF-phenyl H2/H6), 7.52 (t, 3JH,H = 7.4 Hz, 1H, Hpi H7), 7.57 (d, 3J4,5 = 3.1 Hz, 1H, Hpi), 7.69–7.76 (m, 3H, Hpi/HSO2-phenyl H2/H6), 7.78 (d, 3JH,H = 7.3 Hz, 1H, Hpi), 8.00 (s, 1H, NH), 8.09 (d, 3JH,H = 8.8 Hz, 2H, HSO2-phenyl H3/H5) ppm; 13C-NMR (101 MHz, Chloroform-d): δ = 8.4 (CH2CH3), 29.8 (CH2CH3), 111.3 (CHpi), 116.2 (d, 2J3,F = 22 Hz, CHF-phenyl C3/C5), 120.1 (CHpi), 121.7 (CHpi), 122.6 (C), 122.7 (C), 124.3 (C), 124.8 (CHpi), 124.9 (CHpi), 129.1 (CHSO2-phenyl), 129.7 (CHSO2-phenyl), 130.3 (d, 4J1,F = 3 Hz, CF-phenyl C1), 131.3 (d, 3J2,F = 8 Hz, CHF-phenyl C2/C6), 132.6 (C), 137.5 (C), 137.8 (C), 162.3 (d, 1J4,F = 248 Hz, CF-phenyl C4), 171.2 (CONH) ppm; 19F-NMR (376 MHz, (CD3)2SO): δ = −114.3 ppm*, signal of trifluoroacetic acid visible at 76.7 ppm with a molar ratio of product/TFA = 71/1; MS (ESI+): m/z (%) = 311.3 (100) [M + H-SO2NH2COC2H]+, 447.2 (64) [M + H]+; HRMS (ESI/QTOF) m/z: [M + H]+ Calcd for C25H20FN2O3S 447.1173, Found 447.1171; HPLC: 99.9% (tR = 4.79 min; system 3); LogD7.4 HPLC: 2.30 (tR = 13.90 ± 0.03 min).






{kind=link}
{kind=link}
{kind=link}



