
Antileukemic Cell Proliferation of Active Compounds from Kaffir Lime (Citrus hystrix) Leaves

 ,
, 

 , and
, and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Yield of Kaffir Lime Leaf Extracts
2.2. Yield of Fractions and Pure Compounds After Purification Process by Column Chromatography
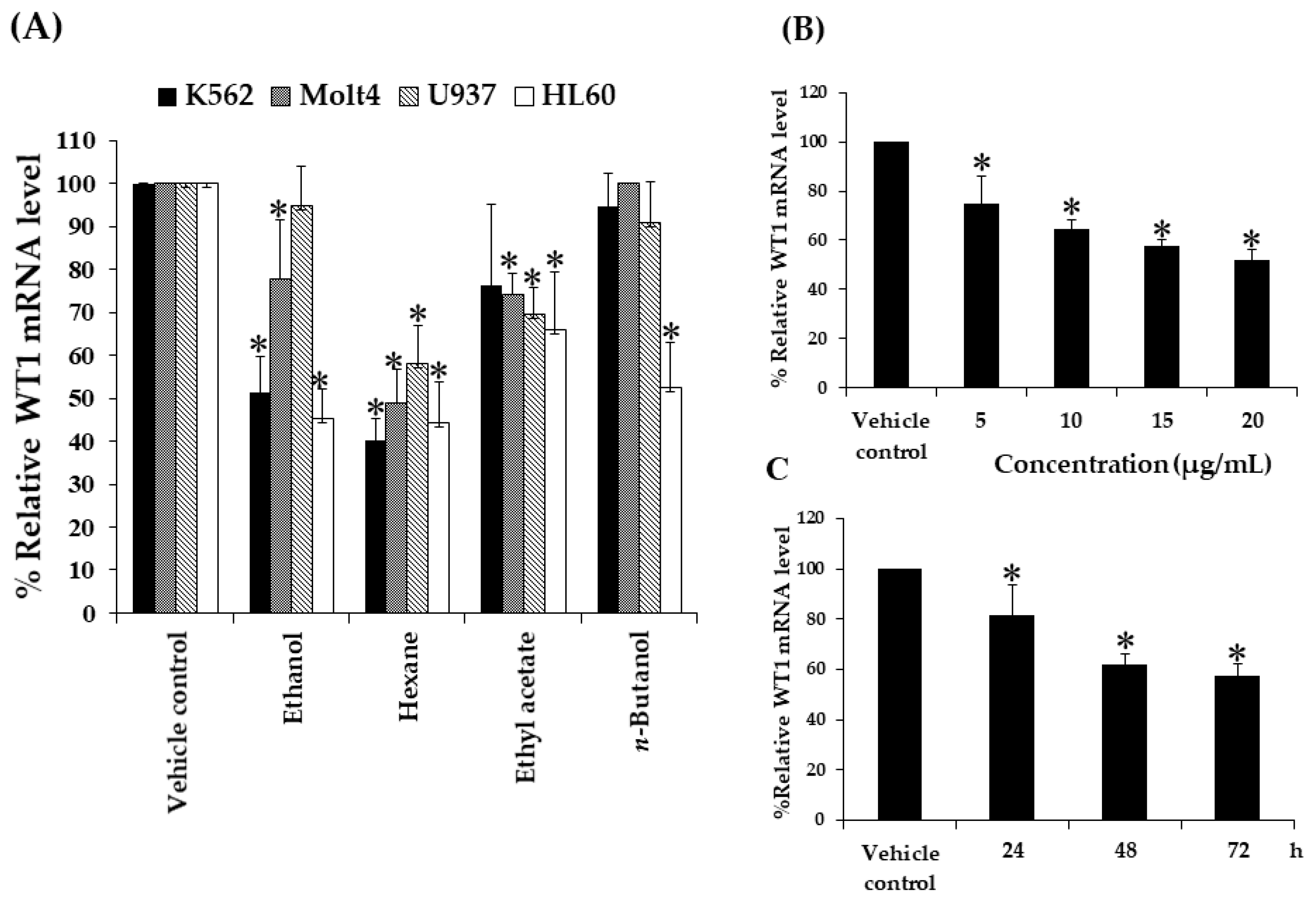
2.3. Effect of the Extracts on WT1 mRNA Levels in K562, Molt4, U937, and HL60 Cell Lines
2.4. Effect of Concentrations and Contact Time of the Extract on WT1 mRNA Levels in K562 Cell Line
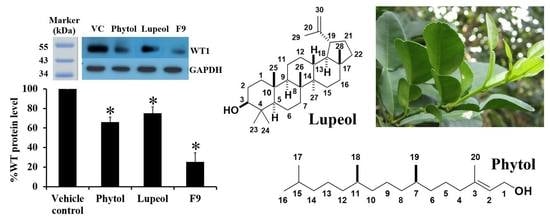
2.5. Effect of the Extracts on WT1 Protein Levels in K562 Cells
2.6. Effect of Concentrations of Extracts on WT1 Protein Levels in K562 Cell Line
2.7. Effect of Contact Time of the Extract on WT1 Protein Levels in K562 Cell Line
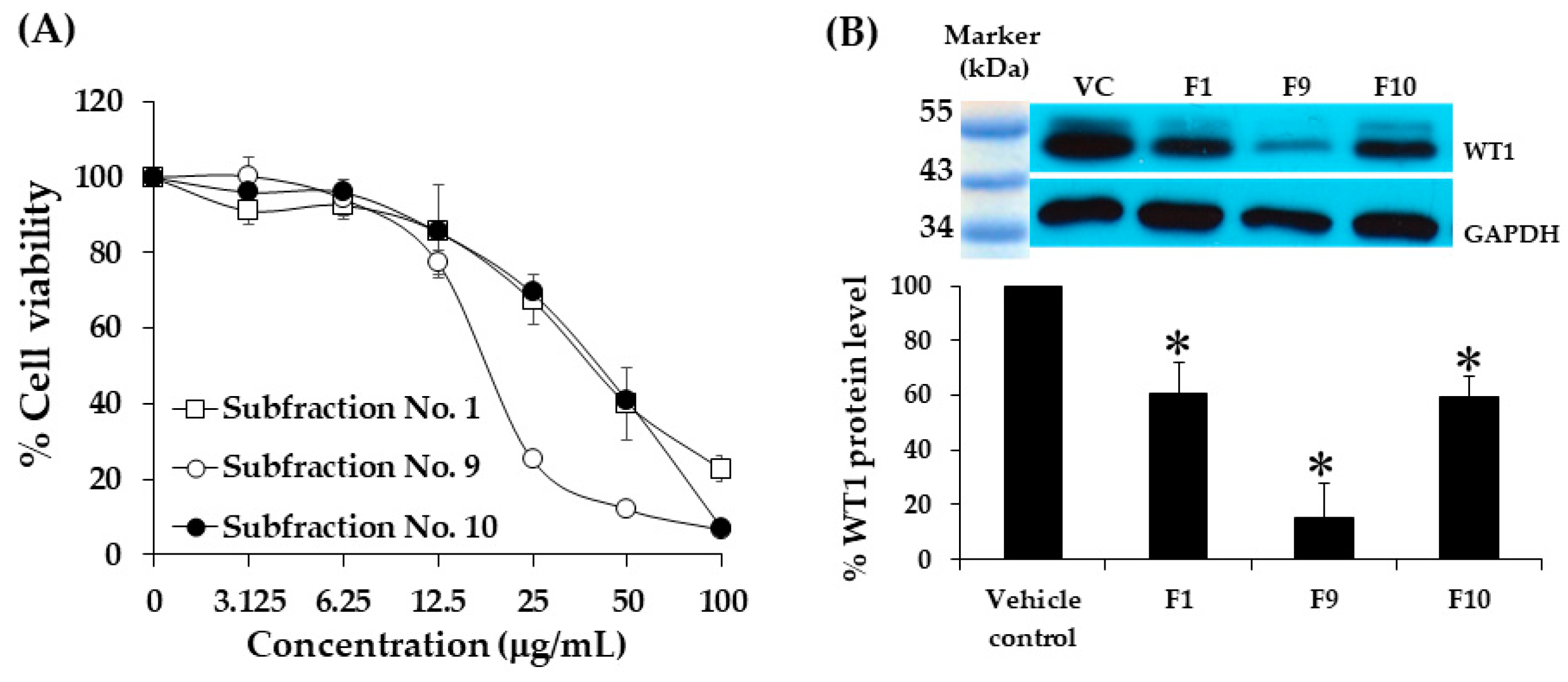
2.8. Cytotoxicity of Subfractions of Hexane Extract on K562 Cells Using MTT Assay
2.9. Effect of Subfraction No. 1, 9, and 10 on WT1 Protein Levels in K562 Cells
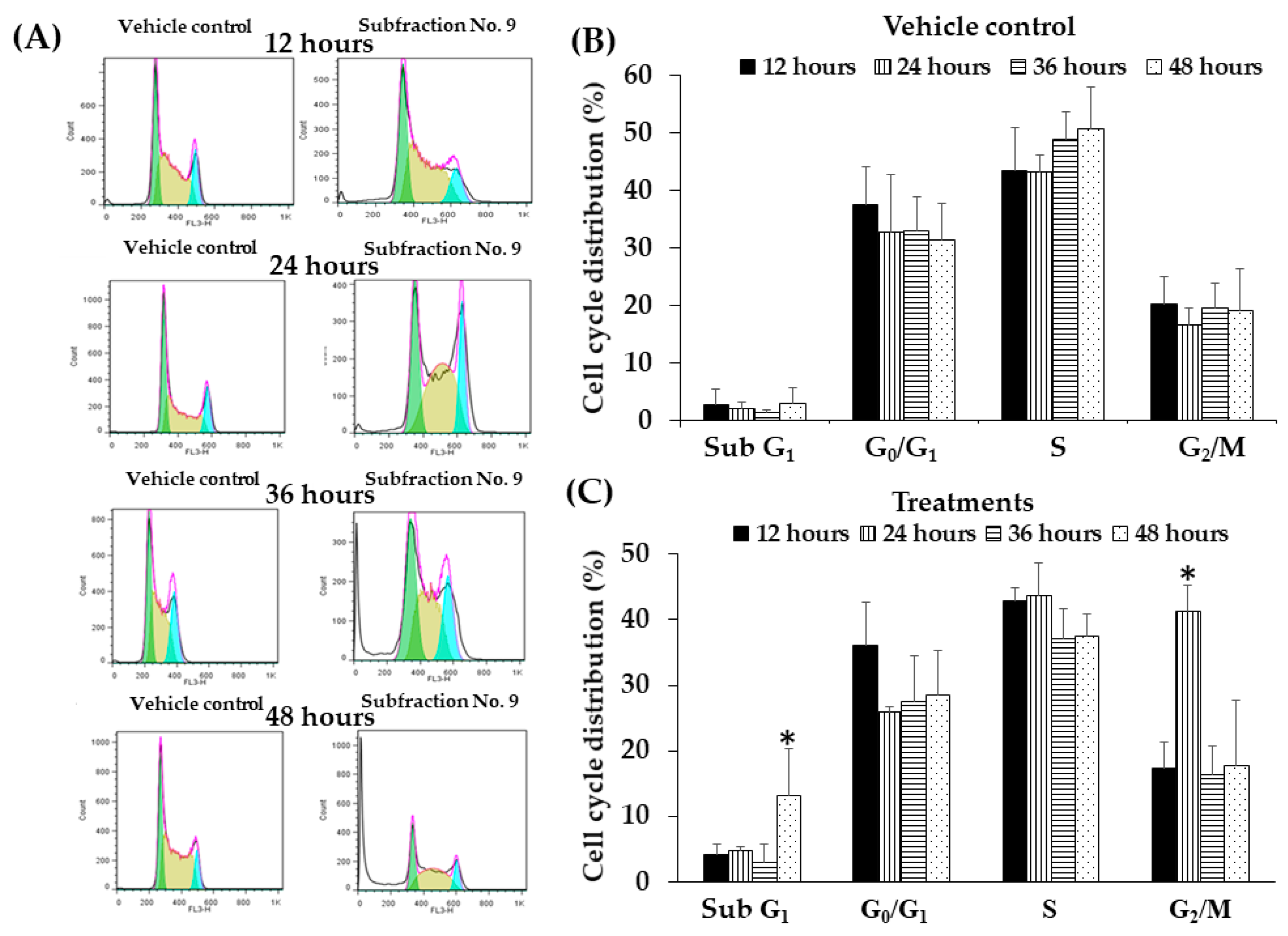
2.10. Effect of F9 on Cell Cycle Distribution in K562 Cell Line by Flow Cytometry
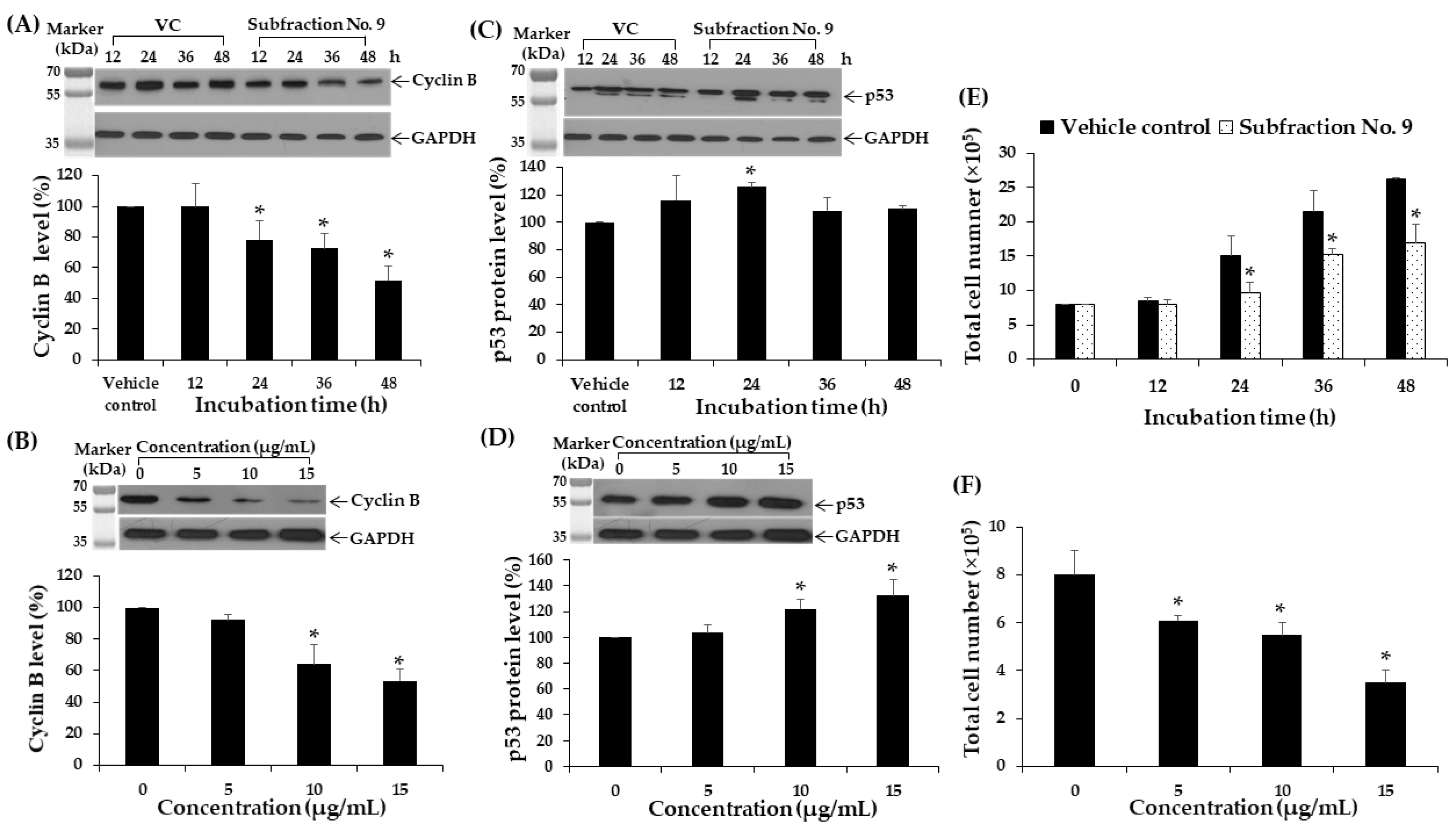
2.11. Effect of F9 on Cyclin B and p53 Protein Expressions in K562 Cell Line
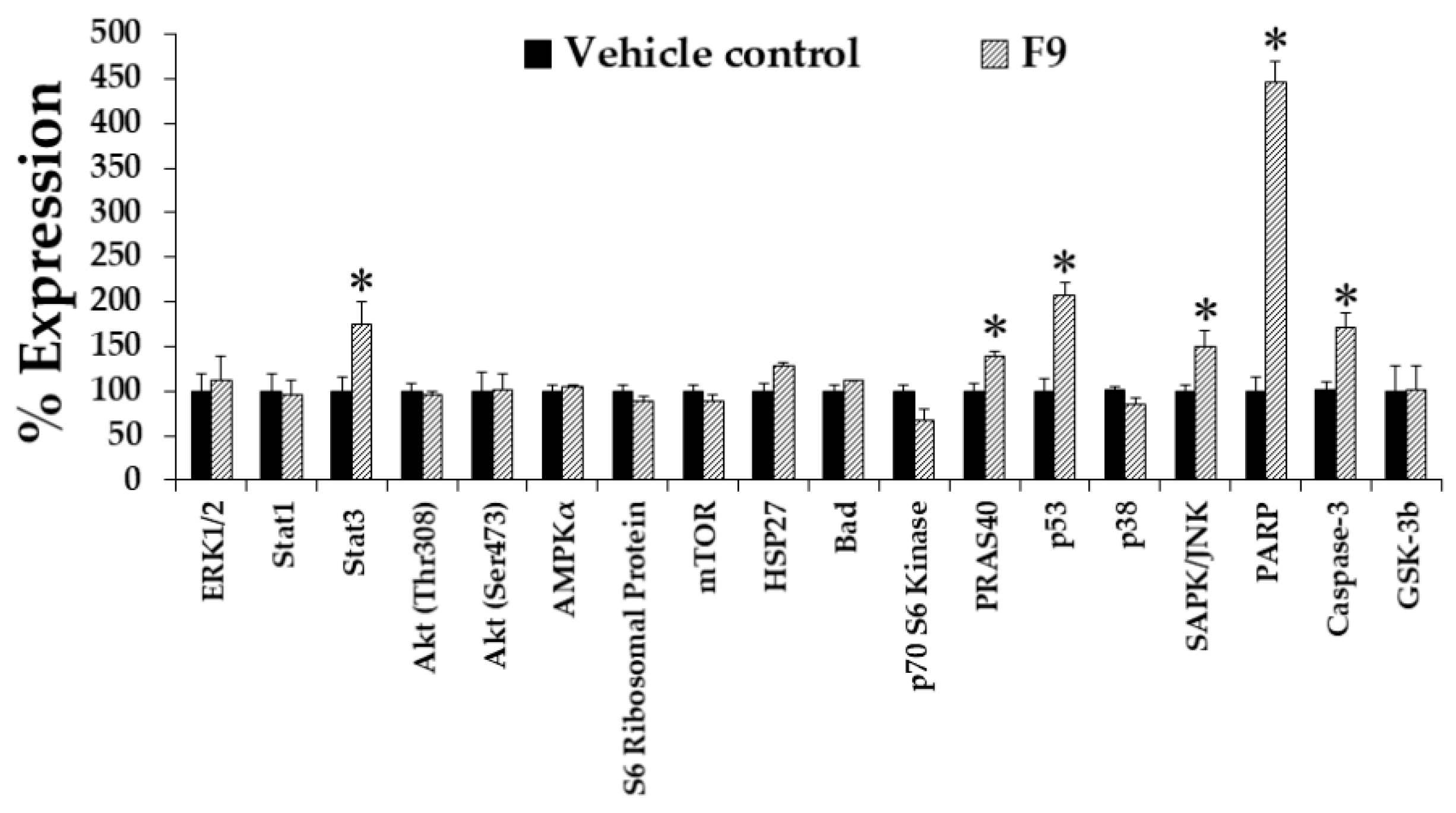
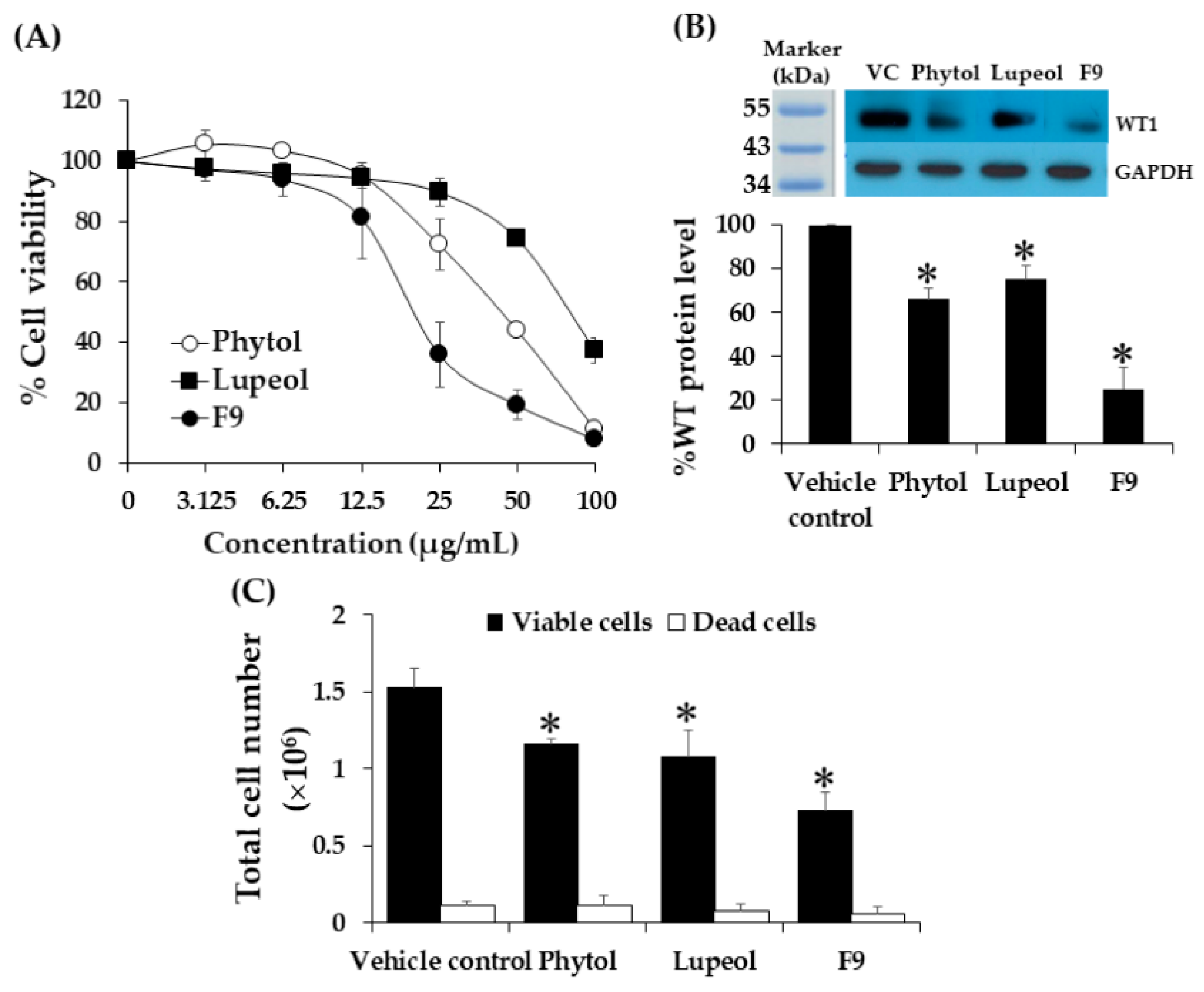
2.12. Effect of Phytol and Lupeol on Cytotoxicity and WT1 Protein Expression in K562 Cells
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Plant Material and Extract Preparation
3.3. Partial Purification by Vacuum Chromatography
3.4. Purification of Compounds from Active Fraction
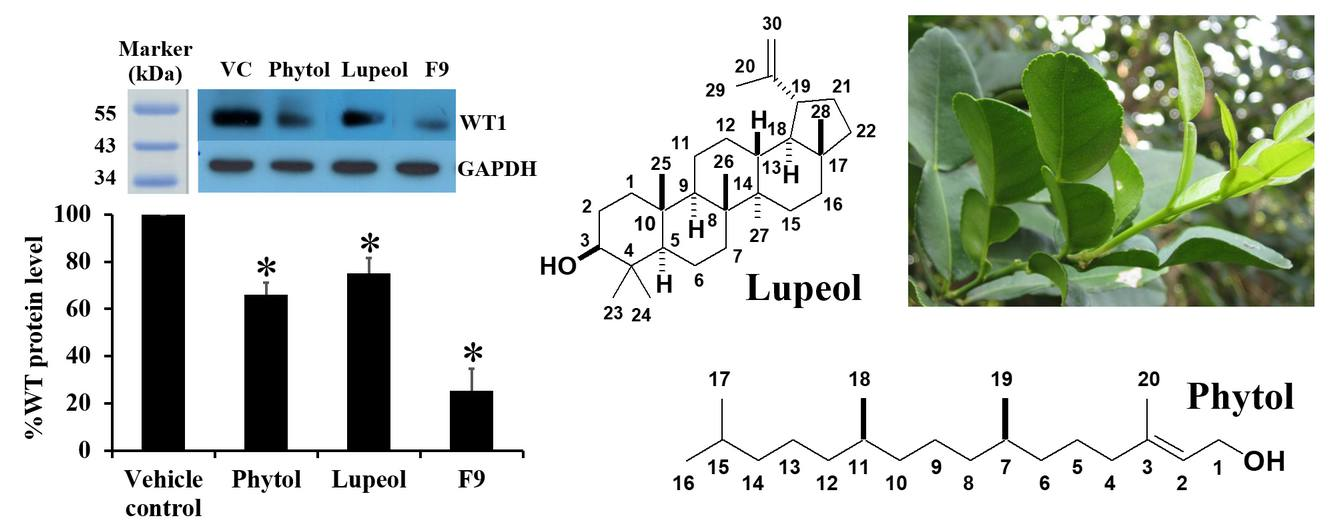
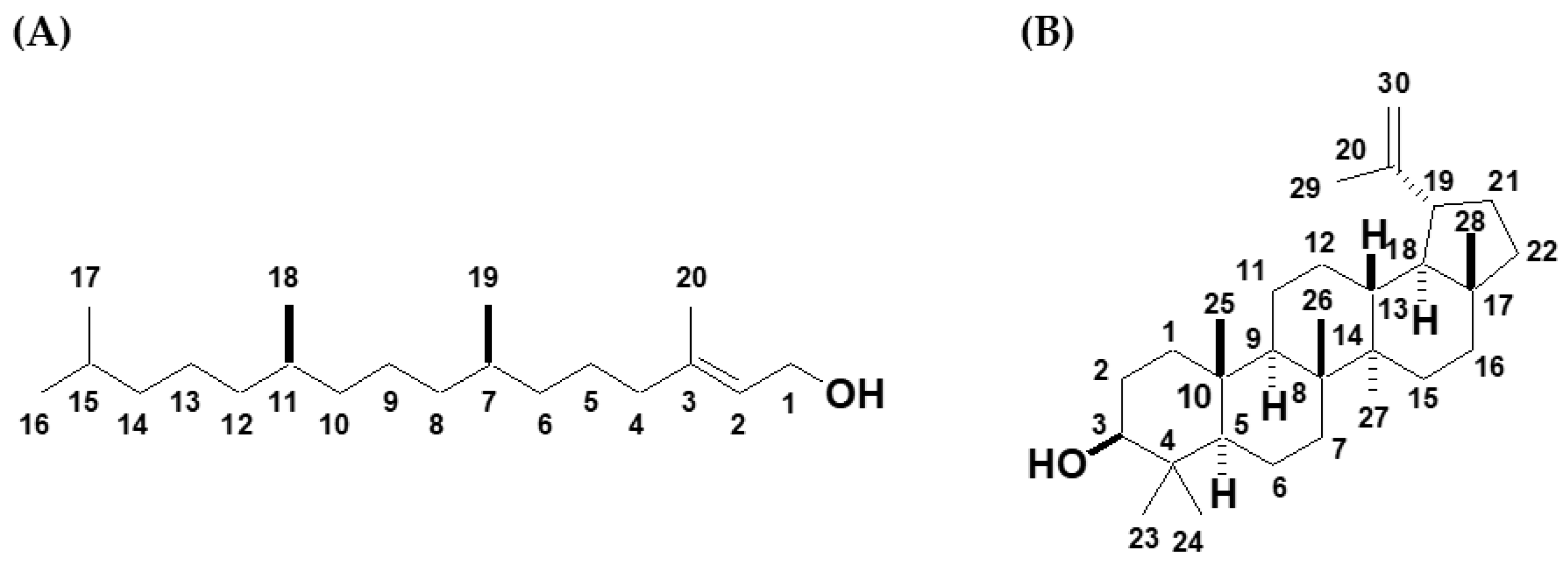
3.5. Characterizations of the Pure Compounds
3.6. Leukemic Cells and Culture Conditions
3.7. MTT Cytotoxicity Assay
3.8. Total RNA Extraction
3.9. cDNA Synthesis
3.10. Real-time RT-PCR
3.11. Protein Extraction and Western Blotting
3.12. Cell Cycle Analysis by Flow Cytometry
3.13. Intracellular Signaling Array
3.14. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Abirami, A.; Nagarani, G.; Siddhuraju, P. The medicinal and nutritional role of underutilized citrus fruit-Citrus hystrix (kaffir lime): a review. Drug Invent. Today. 2014, 6, 1–5. [Google Scholar]
- Lawrence, B.M.; Hogg, J.W.; Terhune, S.J.; Podimuang, V. Constituents of the leaf and peel oils of Citrus hystrix, D.C. Phytochemistry. 1971, 10, 1404–1405. [Google Scholar] [CrossRef]
- Chanthaphon, S.; Chanthachum, S.; Hongpattarakere, T. Antimicrobial activities of essential oils and crude extracts from tropical Citrus spp. against food-related microorganisms. Songklanakarin J. Sci. Tech. 2008, 30, 125–131. [Google Scholar]
- Kasuan, N.; Yunus, M.; Rahiman, M.H.F.; Aris, S.R.S.; Taib, M.N. Essential oil composition of kaffir lime: comparative analysis between controlled steam distillation and hydrodistillation extraction process. In Proceedings of the 2009 IEEE Student Conference on Research and Development (SCOReD), Serdang, Malayasia, 16–18 November 2009; pp. 479–482. [Google Scholar]
- Waikedre, J.; Dugay, A.; Barrachina, I.; Herrenknecht, C.; Cabalion, P.; Fournet, A. Chemical composition and antimicrobial activity of the essential oils from New Caledonian Citrus macroptera and Citrus hystrix. Chem. Biodivers. 2010, 7, 871–877. [Google Scholar] [CrossRef] [PubMed]
- Tachakittirungrod, S.; Okonogi, S.; Chowwanapoonpohn, S. Study on antioxidant activity of certain plants in Thailand: mechanism of antioxidant action of guava leaf extract. Food Chem. 2007, 103, 381–388. [Google Scholar] [CrossRef]
- Jantarach, J.; Thanaboripat, D. The efficacy of ethyl acetate extract of Trichoderma culture broth on growth inhibition and aflatoxin production by Aspergillus flavus IMI 242684. KMITL Sci. Tech. J. 2010, 10, 19–29. [Google Scholar]
- Hutadilok-Towatana, N.; Chaiyamutti, P.; Panthong, K.; Mahabusarakam, W.; Rukachaisirikul, V. Antioxidative and free radical scavenging activities of some plants used in Thai folk medicine. Pharm. Biol. 2006, 44, 221–228. [Google Scholar] [CrossRef]
- Poulose, S.M.; Harris, E.D.; Patil, B.S. Antiproliferative effects of citrus limonoids against human neuroblastoma and colonic adenocarcinoma cells. Nutr. Cancer 2006, 56, 103–112. [Google Scholar] [CrossRef]
- Patil, J.R.; Jayaprakasha, G.; Murthy, K.C.; Tichy, S.E.; Chetti, M.B.; Patil, B.S. Apoptosis-mediated proliferation inhibition of human colon cancer cells by volatile principles of Citrus aurantifolia. Food Chem. 2009, 114, 1351–1358. [Google Scholar] [CrossRef]
- Sugiyama, S.; Umehara, K.; Kuroyanagi, M.; Ueno, A.; Taki, T. Studies on the differentiation inducers of myeloid leukemic cells from Citrus species. Chem. Pharm. Bull. 1993, 41, 714–719. [Google Scholar] [CrossRef] [Green Version]
- Murakami, A.; Nakamura, Y.; Koshimizu, K.; Ohigashi, H. Glyceroglycolipids from Citrus hystrix, a traditional herb in Thailand, potently inhibit the tumor-promoting activity of 12-O-tetradecanoylphorbol 13-acetate in mouse skin. J. Agric. Food Chem. 1995, 43, 2779–2783. [Google Scholar] [CrossRef]
- Manosroi, J.; Dhumtanom, P.; Manosroi, A. Anti-proliferative activity of essential oil extracted from Thai medicinal plants on KB and P388 cell lines. Cancer Lett. 2006, 235, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Ampasavate, C.; Okonogi, S.; Anuchapreeda, S. Cytotoxicity of extracts from fruit plants against leukemic cell lines. Afr. J. Pharm. Pharmacol. 2010, 4, 013–021. [Google Scholar]
- Chueahongthong, F.; Ampasavate, C.; Okonogi, S.; Tima, S.; Anuchapreeda, S. Cytotoxic effects of crude kaffir lime (Citrus hystrix, DC.) leaf fractional extracts on leukemic cell lines. J. Med. Plants Res. 2011, 5, 3097–3105. [Google Scholar]
- Sugiyama, H. Wilms’ tumor gene WT1: its oncogenic function and clinical application. Int. J. Hematol. 2001, 73, 177–187. [Google Scholar] [CrossRef]
- Bergmann, L.; Maurer, U.; Weidmann, E. Wilms tumor gene expression in acute myeloid leukemias. Leuk. Lymphoma. 1997, 25, 435–443. [Google Scholar] [CrossRef]
- Inoue, K.; Sugiyama, H.; Ogawa, H.; Nakagawa, M.; Yamagami, T.; Miwa, H.; Kita, K.; Hiraoka, A.; Masaoka, T.; Nasu, K. WT1 as a new prognostic factor and a new marker for the detection of minimal residual disease in acute leukemia. Blood 1994, 84, 3071–3079. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, L.; Miething, C.; Maurer, U.; Brieger, J.; Karakas, T.; Weidmann, E.; Hoelzer, D. High levels of Wilms′ tumor gene (WT1) mRNA in acute myeloid leukemias are associated with a worse long-term outcome. Blood 1997, 90, 1217–1225. [Google Scholar] [CrossRef]
- Semsri, S.; Krig, S.R.; Kotelawala, L.; Sweeney, C.A.; Anuchapreeda, S. Inhibitory mechanism of pure curcumin on Wilms′ tumor 1 (WT1) gene expression through the PKCα signaling pathway in leukemic K562 cells. FEBS Lett. 2011, 585, 2235–2242. [Google Scholar] [CrossRef] [Green Version]
- Anuchapreeda, S.; Rungrojsakul, M.; Tima, S.; Chiampanichayakul, S.; Krig, S.R. Co-activation of WT1 and AP-1 proteins on WT1 gene promoter to induce WT1 gene expression in K562 cells. Cell. Signal. 2019, 53, 339–347. [Google Scholar] [CrossRef]
- Willstätter, R.; Mayer, E.W.; Hüni, E. Untersuchungen über Chlorophyll. XII. Über Phytol I. Justus Liebigs Ann. Chem. 1911, 378, 73–152. [Google Scholar] [CrossRef] [Green Version]
- Likiernik, A. Ueber das Lupeol. Zeitschrift für physiologische Chemie 1891, 15, 415–425. [Google Scholar] [CrossRef] [Green Version]
- Anuchapreeda, S.; Thanarattanakorn, P.; Sittipreechacharn, S.; Chanarat, P.; Limtrakul, P. Curcumin inhibits WT1 gene expression in human leukemic K562 cells. Acta Pharmacol. Sin. 2006, 27, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Rungrojsakul, M.; Katekunlaphan, T.; Saiai, A.; Ampasavate, C.; Okonogi, S.; Sweeney, C.A.; Anuchapreeda, S. Down-regulatory mechanism of mammea E/BB from Mammea siamensis seed extract on Wilms′ Tumor 1 expression in K562 cells. BMC Complement. Altern. Med. 2016, 16, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerst, G.; Bergold, N.; Viebahn, S.; Gieseke, F.; Kalinova, M.; Trka, J.; Handgretinger, R.; Müller, I. WT1 protein expression in slowly proliferating myeloid leukemic cell lines is scarce throughout the cell cycle with a minimum in G0/G1 phase. Leukemia Res. 2008, 32, 1393–1399. [Google Scholar] [CrossRef] [PubMed]
- Anuchapreeda, S.; Tima, S.; Duangrat, C.; Limtrakul, P. Effect of pure curcumin, demethoxycurcumin, and bisdemethoxycurcumin on WT1 gene expression in leukemic cell lines. Cancer Chemother. Pharmacol. 2008, 62, 585–594. [Google Scholar] [CrossRef]
- Hochedlinger, K.; Wagner, E.F.; Sabapathy, K. Differential effects of JNK1 and JNK2 on signal specific induction of apoptosis. Oncogene 2002, 21, 2441–2445. [Google Scholar] [CrossRef] [Green Version]
- Tournier, C.; Hess, P.; Yang, D.D.; Xu, J.; Turner, T.K.; Nimnual, A.; Bar-Sagi, D.; Jones, S.N.; Flavell, R.A.; Davis, R.J. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 2000, 288, 870–874. [Google Scholar] [CrossRef]
- Dertyasasa, E.D.; Tunjung, W.A.S. Volatile organic compounds of kaffir lime (Citrus hystrix DC.) leaves fractions and their potency as traditional medicine. Biosci. Biotech. Res. Asia. 2017, 14, 1235–1250. [Google Scholar] [CrossRef]
- Tunjung, W.A.S.; Liana, D.; Hidayati, L. Antibacterial activity and composition of crude extracts of kaffir lime (Citrus hystrix DC.) leaves and callus. Proc. Pakistan Acad. Sci. 2018, 55, 45–53. [Google Scholar]
- Pejin, B.; Kojic, V.; Bogdanovic, G. An insight into the cytotoxic activity of phytol at in vitro conditions. Nat. Prod. Res. 2014, 28, 2053–2056. [Google Scholar] [CrossRef] [PubMed]
- Saleem, M. Lupeol, a novel anti-inflammatory and anti-cancer dietary triterpene. Cancer Lett. 2009, 285, 109–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Sample Availability: Samples of the compounds, phytol and lupeol are not available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crude Kaffir Lime Leaf Fractional Extracts | IC20 (µg/mL) (Mean ± SD) | |||
|---|---|---|---|---|
| K562 | Molt4 | U937 | HL60 | |
| Ethanol | 40.9 ± 1.3 | 25.0 ± 3.8 | 10.2 ± 1.4 | 17.0 ± 5.8 |
| Hexane | 13.6 ± 6.9 | 2.8 ± 4.7 | 2.8 ± 3.1 | 3.7 ± 2.0 |
| Ethyl acetate | 11.9 ± 4.4 | 8.0 ± 0.6 | 3.2 ± 2.3 | 8.8 ± 1.4 |
| n-Butanol | 45.4 ± 1.6 | 37.5 ± 2.0 | 18.2 ± 5.3 | 25.0 ± 4.6 |
| Drug. | IC50 Value (Mean ± SD) (µg/mL) | IC20 Value (Mean ± SD) (µg/mL) |
|---|---|---|
| Doxorubicin | 0.80 ± 0.06 | 0.36 ± 0.04 |
| Idarubicin | 0.41 ± 0.04 | 0.09 ± 0.02 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anuchapreeda, S.; Chueahongthong, F.; Viriyaadhammaa, N.; Panyajai, P.; Anzawa, R.; Tima, S.; Ampasavate, C.; Saiai, A.; Rungrojsakul, M.; Usuki, T.; et al. Antileukemic Cell Proliferation of Active Compounds from Kaffir Lime (Citrus hystrix) Leaves. Molecules 2020, 25, 1300. https://doi.org/10.3390/molecules25061300
Anuchapreeda S, Chueahongthong F, Viriyaadhammaa N, Panyajai P, Anzawa R, Tima S, Ampasavate C, Saiai A, Rungrojsakul M, Usuki T, et al. Antileukemic Cell Proliferation of Active Compounds from Kaffir Lime (Citrus hystrix) Leaves. Molecules. 2020; 25(6):1300. https://doi.org/10.3390/molecules25061300
Chicago/Turabian StyleAnuchapreeda, Songyot, Fah Chueahongthong, Natsima Viriyaadhammaa, Pawaret Panyajai, Riki Anzawa, Singkome Tima, Chadarat Ampasavate, Aroonchai Saiai, Methee Rungrojsakul, Toyonobu Usuki, and et al. 2020. "Antileukemic Cell Proliferation of Active Compounds from Kaffir Lime (Citrus hystrix) Leaves" Molecules 25, no. 6: 1300. https://doi.org/10.3390/molecules25061300