Earliest Mechanisms of Dopaminergic Neurons Sufferance in a Novel Slow Progressing Ex Vivo Model of Parkinson Disease in Rat Organotypic Cultures of Substantia Nigra

 ,
,  , and
, and
Abstract
:
1. Introduction
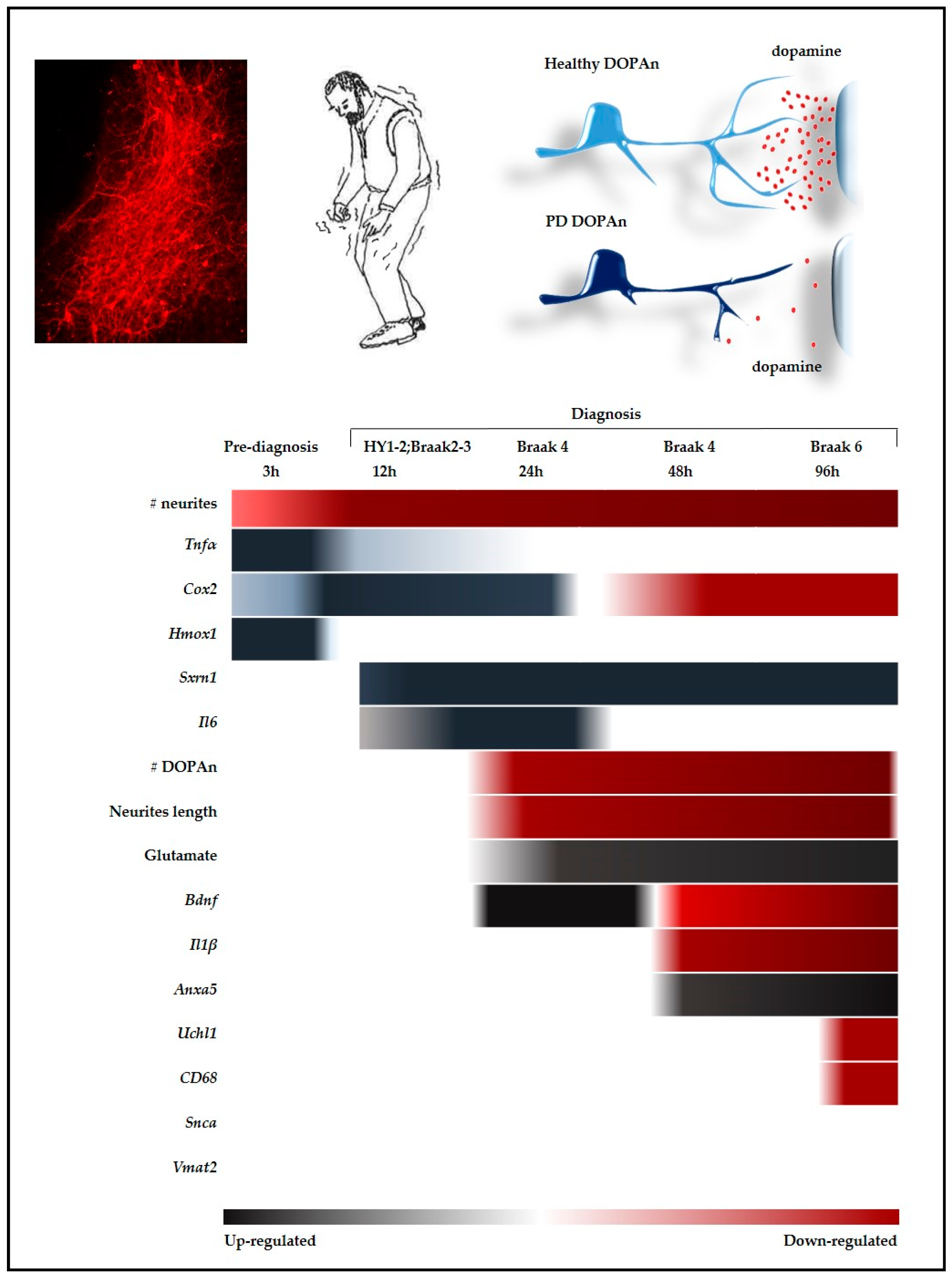
2. Results
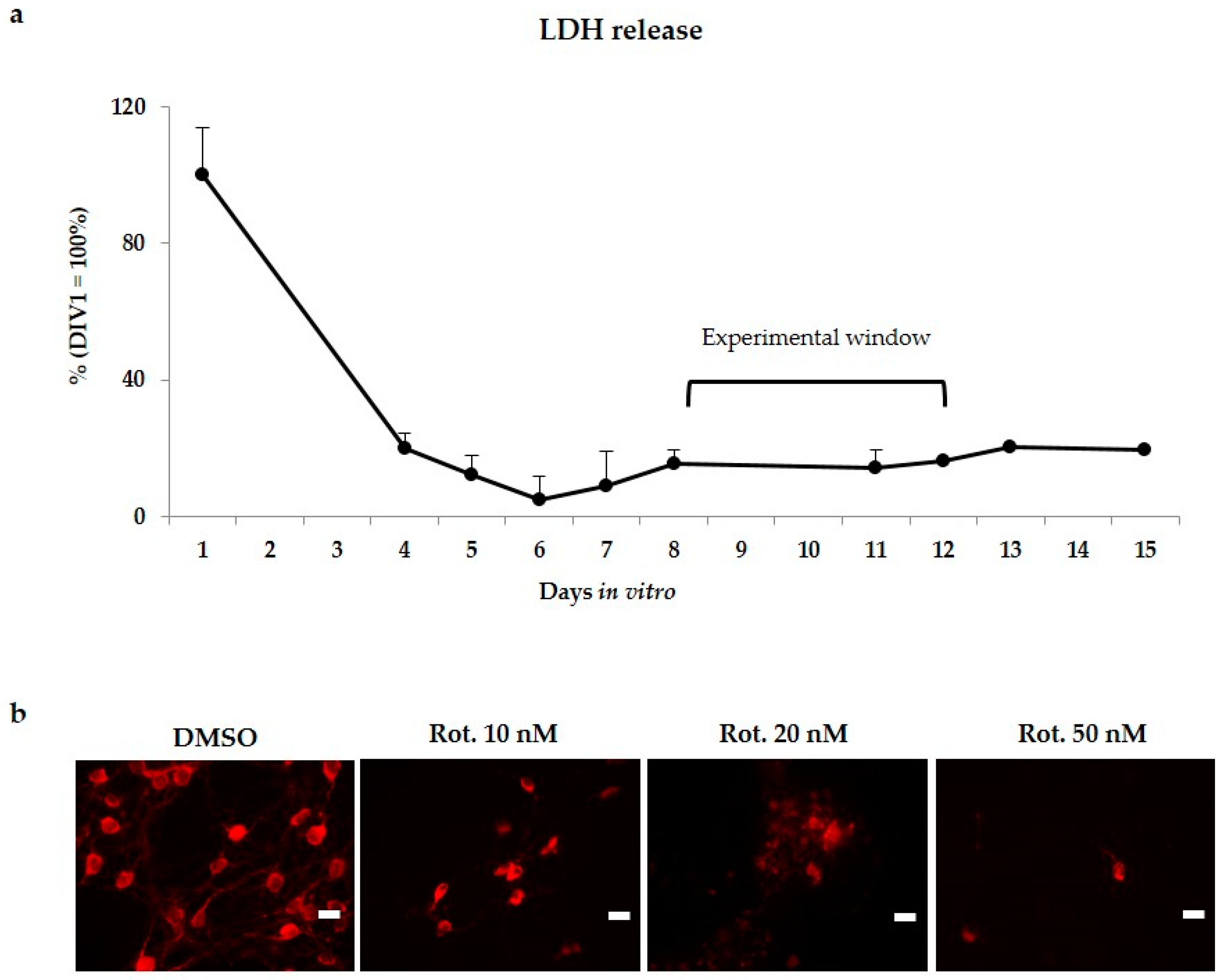
2.1. Model Development and Experimental Plan
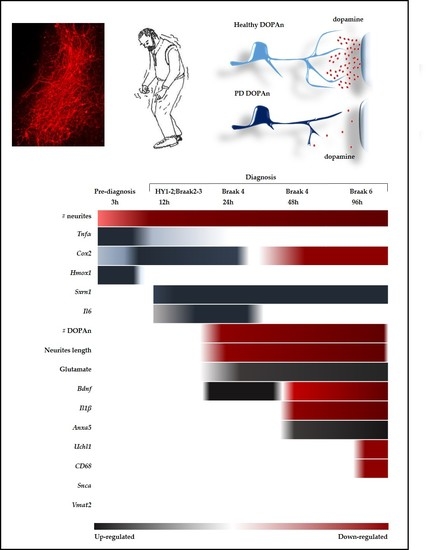
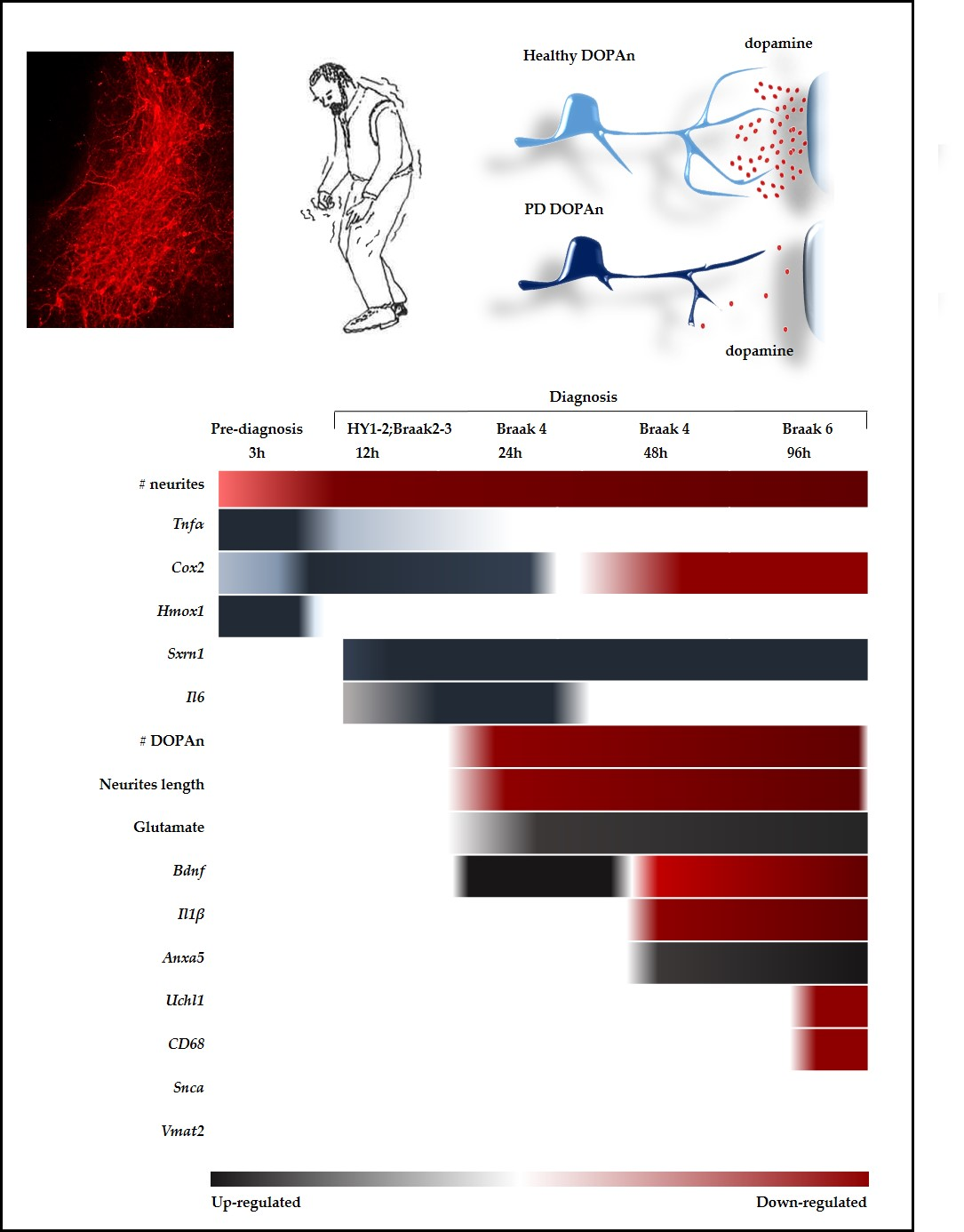
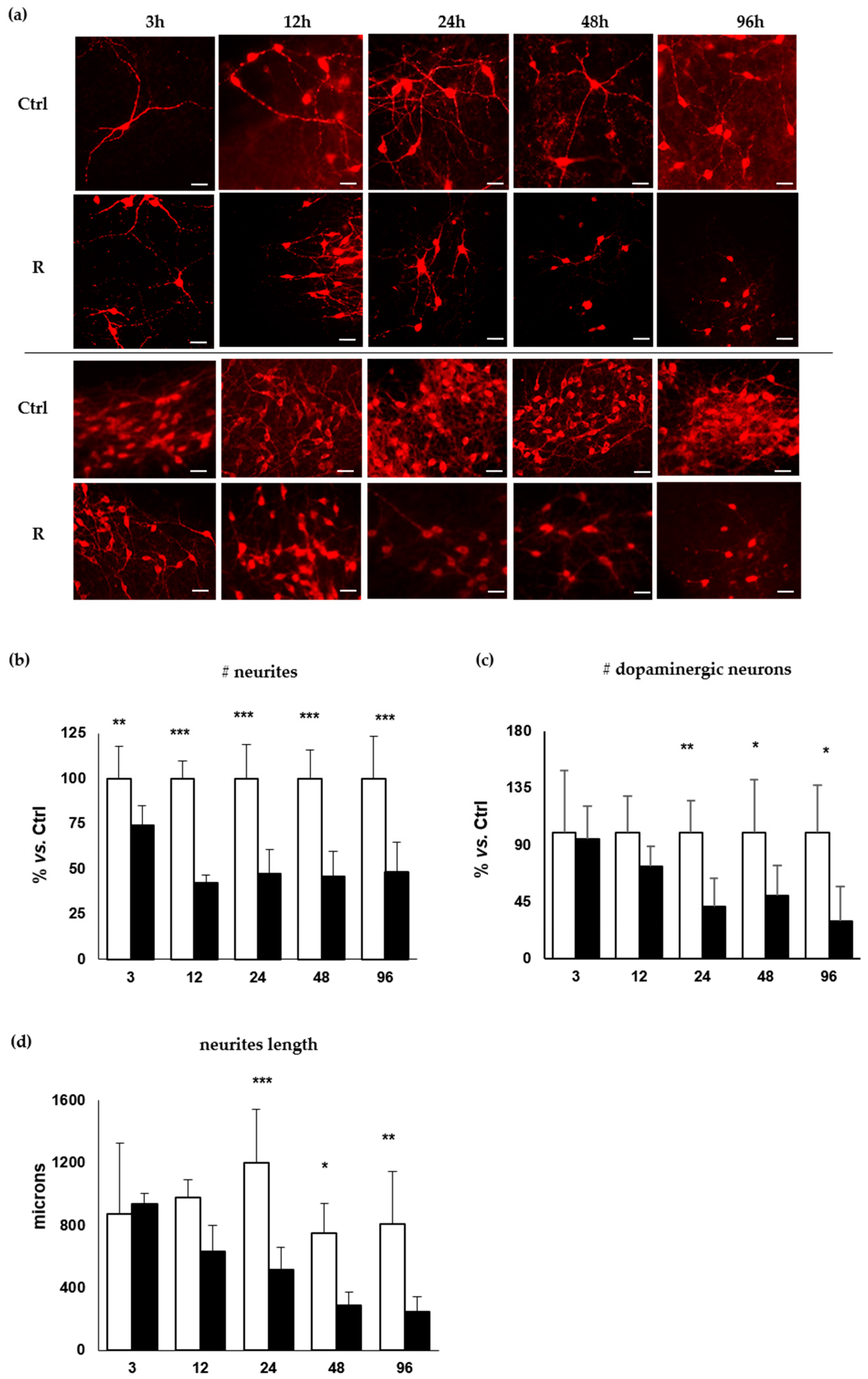
2.2. Morphometric Analysis of the Temporal Degeneration of Dopaminergic Neurons and Agreement with the Clinical Scenario
2.2.1. Decrease of the Number of the DOPAn’s Neurites
2.2.2. Decrease of the Number of the DOPAn
2.2.3. Decrease of the Length of the DOPAn’s Neurites
2.3. Time-Dependent Comparative Evaluation of the Molecular Events Undergoing DOPAn Sufferance and Demise
2.3.1. Real-Time PCR Analysis of Inflammation, Redox Imbalance, Unfolded Proteins Ubiquitination, Vesicles Transport, and Apoptosis; and Quantification of Glutamate in the Medium
2.3.2. Results at 3 h
2.3.3. Results at 12 h
2.3.4. Results at 24 h, 48 h and 96 h
3. Discussion
4. Materials and Methods
4.1. Organotypic Brain Culture Preparation
4.2. Cultures Medium and Treatment
4.3. LDH Test
4.4. Immunofluorescent Staining of Dopaminergic Neurons
4.5. Counting of Dopaminergic Neurons and Neurites
4.6. Glutamate Quantification in Culture Media
4.7. Quantitative Real-Time PCR of Selected Marker Genes
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PD | Parkinson disease |
| DOPAn | Dopaminergic neurons |
| SN | Substantia nigra |
| TH+ | Tyrosine hydroxylase |
| HY | Hohen & Yhar score |
| PET | Positron Emission Tomography |
| Hmox1 | Heme-oxygenase 1 |
| Srnx1 | Sulfiredoxin 1 |
| ROS | Reactive oxygen species |
| Tnfα | Tumor necrosis factor alfa α |
| Cox2 | Cyclo-oxygenase 2 |
| Il6 | Interleukine 6 |
| Il1 β | Interleukine 1β |
| Cd68 | Cluster of differentiation 68, marker of microglia activation |
| Bdnf | Brain-derived neurotrophic factor |
| Snca | Alpha-synuclein (also known as Park1,4) |
| Uchl1 | Ubiquitin carboxy-terminal hydrolase L1 (also known as Park5) |
| Vmat2 | Vesicular monoamine transporter 2 |
| Anxa5 | Annexin5 |
| Glu | Glutamate |
| Gdnf | Glial cell-derived neurotrophic factor |
References
- De Lau, L.M.; Breteler, M.M. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006, 5, 525–535. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Pathoanatomy of Parkinson’s disease. J. Neurol. 2000, 247, II3–II10. [Google Scholar] [CrossRef]
- Shulman, J.M.; De Jager, P.L.; Feany, M.B. Parkinson’s disease: Genetics and pathogenesis. Annu. Rev. Pathol. 2011, 6, 193–222. [Google Scholar] [CrossRef]
- Dickson, D.W. Parkinson’s disease and parkinsonism: Neuropathology. Cold Spring Harb. Perspect. Med. 2012, 2, a009258. [Google Scholar] [CrossRef] [PubMed]
- Dexter, D.T.; Jenner, P. Parkinson disease: From pathology to molecular disease mechanisms. Free Radic. Biol. Med. 2013, 62, 132–144. [Google Scholar] [CrossRef]
- Hwang, O. Role of oxidative stress in Parkinson’s disease. Exp. Neurobiol. 2013, 22, 11–17. [Google Scholar] [CrossRef]
- Antony, P.M.A.; Diederich, N.J.; Krüger, R.; Balling, R. The hallmarks of Parkinson’s disease. FEBS J. 2013, 280, 5981–5993. [Google Scholar] [CrossRef]
- Klemann, C.J.H.M.; Martens, G.J.M.; Sharma, M.; Martens, M.B.; Isacson, O.; Gasser, T.; Visser, J.E.; Poelmans, G. Integrated molecular landscape of Parkinson’s disease. NPJ Parkinson’s Dis. 2017, 3, 14. [Google Scholar] [CrossRef]
- Reeve, A.; Simcox, E.; Turnbull, D. Ageing and Parkinson’s disease: Why is advancing age the biggest risk factor? Ageing Res. Rev. 2014, 14, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F. Excitotoxicity and nitric oxide in Parkinson’s disease pathogenesis. Ann. Neurol. 1998, 44, S110–S114. [Google Scholar] [CrossRef] [PubMed]
- Dauer, W.; Przedborski, S. Parkinson’s disease: Mechanisms and models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef]
- Espay, A.J.; Schwarzschild, M.A.; Tanner, C.M.; Fernandez, H.H.; Simon, D.K.; Leverenz, J.B.; Merola, A.; Chen-Plotkin, A.; Brundin, P.; Kauffman, M.A.; et al. Biomarker-driven phenotyping in Parkinson’s disease: A translational missing link in disease-modifying clinical trials. Mov. Disord. 2017, 32, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Grünblatt, E.; Mandel, S.; Jacob-Hirsch, J.; Zeligson, S.; Amariglo, N.; Rechavi, G.; Li, J.; Ravid, R.; Roggendorf, W.; Riederer, P.; et al. Gene expression profiling of parkinsonian substantia nigra pars compacta; alterations in ubiquitin-proteasome, heat shock protein, iron and oxidative stress regulated proteins, cell adhesion/cellular matrix and vesicle trafficking genes. J. Neural Transm. 2004, 111, 1543–1573. [Google Scholar] [CrossRef]
- Lin, D.; Liang, Y.; Zheng, D.; Chen, Y.; Jing, X.; Lei, M.; Zeng, Z.; Zhou, T.; Wu, X.; Peng, S.; et al. Novel biomolecular information in rotenone-induced cellular model of Parkinson’s disease. Gene 2018, 647, 244–260. [Google Scholar] [CrossRef]
- Saghazadeh, A.; Rezaei, N. MicroRNA machinery in Parkinson’s disease: A platform for neurodegenerative diseases. Expert Rev. Neurother. 2015, 17, 1–27. [Google Scholar] [CrossRef]
- Youdim, M.B.H.; Grünblatt, E.; Levites, Y.; Maor, G.; Mandel, S. Early and late molecular events in neurodegeneration and neuroprotection in Parkinson’s disease MPTP model as assessed by cDNA microarray; the role of iron. Neurotox. Res. 2002, 4, 679–689. [Google Scholar] [CrossRef]
- Yuan, H.; Zhang, Z.-W.; Liang, L.-W.; Shen, Q.; Wang, X.-D.; Ren, S.-M.; Ma, H.-J.; Jiao, S.-J.; Liu, P. Treatment strategies for Parkinson’s disease. Neurosci. Bull. 2010, 26, 66–76. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.; Li, W.-W.; Lu, C.-Z. Histone decacetylase inhibitors prevent mitochondrial fragmentation and elicit early neuroprotection against MPP+. CNS Neurosci. Ther. 2014, 20, 308–316. [Google Scholar] [CrossRef]
- Freitas, M.E.; Fox, S.H. Nondopaminergic treatments for Parkinson’s disease: Current and future prospects. Neurodegener. Dis. Manag. 2016, 6, 249–268. [Google Scholar] [CrossRef]
- Xu, L.; Pu, J. Alpha-synuclein in Parkinson’s disease: From pathogenetic dysfunction to potential clinical application. Parkinsons Dis. 2016, 2016, 1720621. [Google Scholar]
- Perry, V.H. Innate inflammation in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009373. [Google Scholar] [CrossRef]
- Mandel, S.; Grünblatt, E.; Maor, G.; Youdim, M.B.H. Early and late gene changes in MPTP mice model of Parkinson’s disease employing cDNA microarray. Neurochem. Res. 2002, 27, 1231–1243. [Google Scholar] [CrossRef]
- Hawkes, C.H.; Del Tredici, K.; Braak, H. A timeline for Parkinson’s disease. Parkinsonism Relat. Disord. 2010, 16, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Jenner, P.; Clark, J.B.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet 1989, 1, 1269. [Google Scholar] [CrossRef]
- Sherer, T.B.; Betarbet, R.; Testa, C.M.; Seo, B.B.; Richardson, J.R.; Kim, J.H.; Miller, G.W.; Yagi, T.; Matsuno-Yagi, A.; Greenamyre, J.T. Mechanism of toxicity in rotenone models of Parkinson’s disease. J. Neurosci. 2003, 23, 10756–10764. [Google Scholar] [CrossRef]
- Tanner, C.M.; Kamel, F.; Ross, G.W.; Hoppin, J.A.; Goldman, S.M.; Korell, M.; Marras, C.; Bhudhikanok, G.S.; Kasten, M.; Chade, A.R.; et al. Rotenone, paraquat, and Parkinson’s disease. Environ. Health Perspect. 2011, 119, 866–872. [Google Scholar] [CrossRef]
- Johnosn, M.E.; Bobrovskaya, L. An update on the rotenone models of Parkinson’s disease: Their ability to reproduce the features of clinical disease and model gene–environment interactions. NeuroToxicology 2015, 46, 101–116. [Google Scholar] [CrossRef]
- Blesa, J.; Przedborski, S. Parkinson’s disease: Animal models and dopaminergic cell vulnerability. Front. Neuroanat. 2014, 8, 155. [Google Scholar] [CrossRef]
- Cheng, H.-C.; Ulane, C.M.; Burke, R.E. Clinical progression in Parkinson disease and the neurobiology of axons. Ann. Neurol. 2010, 67, 715–725. [Google Scholar] [CrossRef]
- Rocha, N.P.; de Miranda, A.S.; Teixeira, A.L. Insights into neuroinflammation in Parkinson’s disease: From biomarkers to anti-inflammatory based therapies. BioMed Res. Int. 2015, 2015, 628192. [Google Scholar] [CrossRef] [PubMed]
- Hoehn, M.M.; Yahr, M.D. Parkinsonism: Onset, progression and mortality. Neurology 1967, 17, 427–442. [Google Scholar] [CrossRef]
- Bhidayasiri, R.; Tarsy, D. Parkinson’s disease: Hoehn and Yahr scale. In Movement Disorders: A Video Atlas; Bhidayasiri, R., Tarsy, D., Eds.; Current Clinical Neurology; Humana Press: Totowa, NJ, USA, 2012; pp. 4–5. ISBN 978-1-60327-426-5. [Google Scholar]
- Fazio, P.; Svenningsson, P.; Cselényi, Z.; Halldin, C.; Farde, L.; Varrone, A. Nigrostriatal dopamine transporter availability in early Parkinson’s disease. Mov. Disord. 2018, 33, 592–599. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.I.; Jansen Steur, E.N.H.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Liu, L.-X.; Du, D.; Zheng, T.; Fang, Y.; Chen, Y.-S.; Yi, H.-L.; He, Q.-Y.; Gao, D.-W.; Shi, Q.-L. Detecting dopaminergic neuronal degeneration using diffusion tensor imaging in a rotenone-induced rat model of Parkinson’s disease: Fractional anisotropy and mean diffusivity values. Neural Regen. Res. 2017, 12, 1485–1491. [Google Scholar] [CrossRef]
- Dijkstra, A.A.; Voorn, P.; Berendse, H.W.; Groenewegen, H.J.; Netherlands Brain Bank; Rozemuller, A.J.M.; van de Berg, W.D.J. Stage-dependent nigral neuronal loss in incidental lewy body and Parkinson’s disease. Mov. Disord. 2014, 29, 1244–1251. [Google Scholar] [CrossRef] [PubMed]
- Blesa, J.; Trigo-Damas, I.; Quiroga-Varela, A.; Jackson-Lewis, V.R. Oxidative stress and Parkinson’s disease. Front. Neuroanat. 2015, 9, 91. [Google Scholar] [CrossRef]
- Mateo, I.; Infante, J.; Sánchez-Juan, P.; García-Gorostiaga, I.; Rodríguez-Rodríguez, E.; Vázquez-Higuera, J.L.; Berciano, J.; Combarros, O. Serum heme oxygenase-1 levels are increased in Parkinson’s disease but not in Alzheimer’s disease. Acta Neurol. Scand. 2010, 121, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell. Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [PubMed]
- McMahon, M.; Ding, S.; Acosta-Jimenez, L.P.; Frangova, T.G.; Henderson, C.J.; Wolf, C.R. Measuring physiological responses to stressors using a novel Hmox1 reporter mouse. bioRxiv 2017, 098467. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.-C.; Fan, C.-W.; Tseng, W.-K.; Chein, H.-P.; Hsieh, T.-Y.; Chen, J.-R.; Hwang, C.-C.; Hua, C.-C. The Ratio of Hmox1/Nrf2 mRNA Level in the Tumor Tissue Is a Predictor of Distant Metastasis in Colorectal Cancer. Available online: https://www.hindawi.com/journals/dm/2016/8143465/abs/ (accessed on 29 April 2019).
- Li, Q.; Yu, S.; Wu, J.; Zou, Y.; Zhao, Y. Sulfiredoxin-1 protects PC12 cells against oxidative stress induced by hydrogen peroxide. J. Neurosci. Res. 2013, 91, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, Y.; Yu, S.; Wu, J.; Chen, Y.; Zhao, Y. Sulfiredoxin-1 exerts anti-apoptotic and neuroprotective effects against oxidative stress-induced injury in rat cortical astrocytes following exposure to oxygen-glucose deprivation and hydrogen peroxide. Int. J. Mol. Med. 2015, 36, 43–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escobar, J.; Cubells, E.; Enomoto, M.; Quintas, G.; Kuligowski, J.; Martinez-Fernandez de la Camara, C.; Torres-Cuevas, I.; Sastre, J.; Belik, J.; Vento, M. Prolonging in utero-like oxygenation after birth diminishes oxidative stress in the lung and brain of mice pups. Redox Biol. 2013, 1, 297–303. [Google Scholar] [CrossRef]
- Kouchaki, E.; Kakhaki, R.D.; Tamtaji, O.R.; Dadgostar, E.; Behnam, M.; Nikoueinejad, H.; Akbari, H. Increased serum levels of TNF-α and decreased serum levels of IL-27 in patients with Parkinson disease and their correlation with disease severity. Clin. Neurol. Neurosurg. 2018, 166, 76–79. [Google Scholar] [CrossRef]
- Menza, M.; Dobkin, R.D.; Marin, H.; Mark, M.H.; Gara, M.; Bienfait, K.; Dicke, A.; Kusnekov, A. The role of inflammatory cytokines in cognition and other non-motor symptoms of Parkinson’s disease. Psychosomatics 2010, 51, 474–479. [Google Scholar] [PubMed]
- Rocha, N.P.; Teixeira, A.L.; Scalzo, P.L.; Barbosa, I.G.; de Sousa, M.S.; Morato, I.B.; Vieira, E.L.M.; Christo, P.P.; Palotás, A.; Reis, H.J. Plasma levels of soluble tumor necrosis factor receptors are associated with cognitive performance in Parkinson’s disease. Mov. Disord. 2014, 29, 527–531. [Google Scholar] [CrossRef]
- Chen, H.; Jacobs, E.; Schwarzschild, M.A.; McCullough, M.L.; Calle, E.E.; Thun, M.J.; Ascherio, A. Nonsteroidal antiinflammatory drug use and the risk for Parkinson’s disease. Ann. Neurol. 2005, 58, 963–967. [Google Scholar] [CrossRef]
- Gao, X.; Chen, H.; Schwarzschild, M.A.; Ascherio, A. Use of ibuprofen and risk of Parkinson disease. Neurology 2011, 76, 863–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassani, T.B.; Vital, M.A.B.F.; Rauh, L.K. Neuroinflammation in the pathophysiology of Parkinson’s disease and therapeutic evidence of anti-inflammatory drugs. Arq. Neuro Psiquiatr. 2015, 73, 616–623. [Google Scholar] [CrossRef] [Green Version]
- Teismann, P.; Tieu, K.; Choi, D.-K.; Wu, D.-C.; Naini, A.; Hunot, S.; Vila, M.; Jackson-Lewis, V.; Przedborski, S. Cyclooxygenase-2 is instrumental in Parkinson’s disease neurodegeneration. Proc. Natl. Acad. Sci. USA 2003, 100, 5473–5478. [Google Scholar] [CrossRef]
- Pereira, J.R.; Santos, L.V.D.; Santos, R.M.S.; Campos, A.L.F.; Pimenta, A.L.; de Oliveira, M.S.; Bacheti, G.G.; Rocha, N.P.; Teixeira, A.L.; Christo, P.P.; et al. IL-6 serum levels are elevated in Parkinson’s disease patients with fatigue compared to patients without fatigue. J. Neurol. Sci. 2016, 370, 153–156. [Google Scholar] [CrossRef] [PubMed]
- More, S.V.; Kumar, H.; Kim, I.S.; Song, S.-Y.; Choi, D.-K. Cellular and molecular mediators of neuroinflammation in the pathogenesis of Parkinson’s disease. Mediat. Inflamm. 2013, 2013, 952375. [Google Scholar] [CrossRef]
- Doorn, K.J.; Moors, T.; Drukarch, B.; van de Berg, W.D.; Lucassen, P.J.; van Dam, A.-M. Microglial phenotypes and toll-like receptor 2 in the substantia nigra and hippocampus of incidental lewy body disease cases and Parkinson’s disease patients. Acta Neuropathol. Commun. 2014, 2, 90. [Google Scholar]
- Doorn, K.J.; Goudriaan, A.; Blits-Huizinga, C.; Bol, J.G.J.M.; Rozemuller, A.J.; Hoogland, P.V.J.M.; Lucassen, P.J.; Drukarch, B.; van de Berg, W.D.J.; van Dam, A.-M. Increased amoeboid microglial density in the olfactory bulb of Parkinson’s and Alzheimer’s patients. Brain Pathol. 2014, 24, 152–165. [Google Scholar] [CrossRef]
- Howells, D.W.; Porritt, M.J.; Wong, J.Y.; Batchelor, P.E.; Kalnins, R.; Hughes, A.J.; Donnan, G.A. Reduced BDNF mRNA expression in the Parkinson’s disease substantia nigra. Exp. Neurol. 2000, 166, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.; Peppe, A.; Carlesimo, G.A.; Zabberoni, S.; Scalici, F.; Caltagirone, C.; Angelucci, F. Brain-derived neurotrophic factor serum levels correlate with cognitive performance in Parkinson’s disease patients with mild cognitive impairment. Front. Behav. Neurosci. 2015, 9, 253. [Google Scholar] [CrossRef]
- Scalzo, P.; Kümmer, A.; Bretas, T.L.; Cardoso, F.; Teixeira, A.L. Serum levels of brain-derived neurotrophic factor correlate with motor impairment in Parkinson’s disease. J. Neurol. 2010, 257, 540–545. [Google Scholar] [CrossRef]
- Kumar, A.; Tamjar, J.; Waddell, A.D.; Woodroof, H.I.; Raimi, O.G.; Shaw, A.M.; Peggie, M.; Muqit, M.M.; van Aalten, D.M. Structure of PINK1 and mechanisms of Parkinson’s disease-associated mutations. Elife 2017, 6, e29985. [Google Scholar] [CrossRef]
- Locascio, J.J.; Eberly, S.; Liao, Z.; Liu, G.; Hoesing, A.N.; Duong, K.; Trisini-Lipsanopoulos, A.; Dhima, K.; Hung, A.Y.; Flaherty, A.W.; et al. Association between α-synuclein blood transcripts and early, neuroimaging-supported Parkinson’s disease. Brain 2015, 138, 2659–2671. [Google Scholar] [CrossRef] [Green Version]
- Dumitriu, A.; Moser, C.; Hadzi, T.C.; Williamson, S.L.; Pacheco, C.D.; Hendricks, A.E.; Latourelle, J.C.; Wilk, J.B.; DeStefano, A.L.; Myers, R.H. Postmortem Interval Influences α-Synuclein Expression in Parkinson Disease Brain. Available online: https://www.hindawi.com/journals/pd/2012/614212/ (accessed on 29 April 2019).
- Marques, O.; Outeiro, T.F. Alpha-synuclein: From secretion to dysfunction and death. Cell Death Dis. 2012, 3, e350. [Google Scholar] [CrossRef] [PubMed]
- McNaught, K.S.; Jenner, P. Proteasomal function is impaired in substantia nigra in Parkinson’s disease. Neurosci. Lett. 2001, 297, 191–194. [Google Scholar] [CrossRef]
- Takami, Y.; Nakagami, H.; Morishita, R.; Katsuya, T.; Cui, T.-X.; Ichikawa, T.; Saito, Y.; Hayashi, H.; Kikuchi, Y.; Nishikawa, T.; et al. Ubiquitin carboxyl-terminal hydrolase L1, a novel deubiquitinating enzyme in the vasculature, attenuates NF-κB activation. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2184–2190. [Google Scholar] [CrossRef]
- Carrieri, C.; Forrest, A.R.R.; Santoro, C.; Persichetti, F.; Carninci, P.; Zucchelli, S.; Gustincich, S. Expression analysis of the long non-coding RNA antisense to Uchl1 (AS Uchl1) during dopaminergic cells’ differentiation in vitro and in neurochemical models of Parkinson’s disease. Front. Cell. Neurosci. 2015, 9, 114. [Google Scholar] [CrossRef] [PubMed]
- Lohr, K.M.; Miller, G.W. VMAT2 and Parkinson’s disease: Harnessing the dopamine vesicle. Expert Rev. Neurother. 2014, 14, 1115–1117. [Google Scholar] [CrossRef]
- Osherovich, L. Priming the PD pump. SciBX Sci. Bus. Exch. 2014, 7. [Google Scholar] [CrossRef]
- Xue, G.; Hao, L.-Q.; Ding, F.-X.; Mei, Q.; Huang, J.-J.; Fu, C.-G.; Yan, H.-L.; Sun, S.-H. Expression of annexin a5 is associated with higher tumor stage and poor prognosis in colorectal adenocarcinomas. J. Clin. Gastroenterol. 2009, 43, 831–837. [Google Scholar] [CrossRef] [PubMed]
- Sonsalla, P.K.; Albers, D.S.; Zeevalk, G.D. Role of glutamate in neurodegeneration of dopamine neurons in several animal models of Parkinsonism. Amino Acids 1998, 14, 69–74. [Google Scholar] [CrossRef]
- Morales, I.; Sabate, M.; Rodriguez, M. Striatal glutamate induces retrograde excitotoxicity and neuronal degeneration of intralaminar thalamic nuclei: Their potential relevance for Parkinson’s disease. Eur. J. Neurosci. 2013, 38, 2172–2182. [Google Scholar] [CrossRef]
- Schipper, H.M.; Liberman, A.; Stopa, E.G. Neural heme oxygenase-1 expression in idiopathic Parkinson’s disease. Exp. Neurol. 1998, 150, 60–68. [Google Scholar] [CrossRef]
- Moreau, C.; Duce, J.A.; Rascol, O.; Devedjian, J.-C.; Berg, D.; Dexter, D.; Cabantchik, Z.I.; Bush, A.I.; Devos, D. FAIRPARK-II study group iron as a therapeutic target for Parkinson’s disease. Mov. Disord. 2018, 33, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Qaisiya, M.; Coda Zabetta, C.D.; Bellarosa, C.; Tiribelli, C. Bilirubin mediated oxidative stress involves antioxidant response activation via Nrf2 pathway. Cell Signal. 2014, 26, 512–520. [Google Scholar] [CrossRef]
- Hung, S.-Y.; Liou, H.-C.; Kang, K.-H.; Wu, R.-M.; Wen, C.-C.; Fu, W.-M. Overexpression of heme oxygenase-1 protects dopaminergic neurons against 1-Methyl-4-Phenylpyridinium-Induced neurotoxicity. Mol. Pharmacol. 2008, 74, 1564–1575. [Google Scholar] [CrossRef]
- Soriano, F.X.; Baxter, P.; Murray, L.M.; Sporn, M.B.; Gillingwater, T.H.; Hardingham, G.E. Transcriptional regulation of the AP-1 and Nrf2 target gene sulfiredoxin. Mol. Cells 2009, 27, 279–282. [Google Scholar] [CrossRef] [PubMed]
- Abbas, K.; Breton, J.; Planson, A.-G.; Bouton, C.; Bignon, J.; Seguin, C.; Riquier, S.; Toledano, M.B.; Drapier, J.-C. Nitric oxide activates an Nrf2/sulfiredoxin antioxidant pathway in macrophages. Free Radic. Biol. Med. 2011, 51, 107–114. [Google Scholar] [CrossRef]
- Sunico, C.R.; Sultan, A.; Nakamura, T.; Dolatabadi, N.; Parker, J.; Shan, B.; Han, X.; Yates, J.R.; Masliah, E.; Ambasudhan, R.; et al. Role of sulfiredoxin as a peroxiredoxin-2 denitrosylase in human iPSC-derived dopaminergic neurons. Proc. Natl. Acad. Sci. USA 2016, 113, E7564–E7571. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; He, Z.; Guo, J.; Li, Z.; Wang, X.; Yang, C.; Cui, X. Sulfiredoxin-1 protects against simulated ischaemia/reperfusion injury in cardiomyocyte by inhibiting PI3K/AKT-regulated mitochondrial apoptotic pathways. Biosci. Rep. 2016, 36, e00325. [Google Scholar] [CrossRef] [PubMed]
- Tansey, M.G.; Goldberg, M.S. Neuroinflammation in Parkinson’s disease: Its role in neuronal death and implications for therapeutic intervention. Neurobiol. Dis. 2010, 37, 510–518. [Google Scholar] [CrossRef]
- Tansey, M.G.; McCoy, M.K.; Frank-Cannon, T.C. Neuroinflammatory mechanisms in Parkinson’s disease: Potential environmental triggers, pathways, and targets for early therapeutic intervention. Exp. Neurol. 2007, 208, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Jangir, D.K.; Verma, G.; Shekhar, S.; Hanpude, P.; Kumar, S.; Kumari, R.; Singh, N.; Sarovar Bhavesh, N.; Ranjan Jana, N.; et al. S-nitrosylation of UCHL1 induces its structural instability and promotes α-synuclein aggregation. Sci. Rep. 2017, 7, 44558. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, A.M.; O’Keeffe, G.W. Neurotrophic factor therapy for Parkinson’s disease: Past, present and future. Neural Regen. Res. 2016, 11, 205–207. [Google Scholar] [CrossRef] [PubMed]
- Stahl, K.; Mylonakou, M.N.; Skare, Ø.; Amiry-Moghaddam, M.; Torp, R. Cytoprotective effects of growth factors: BDNF more potent than GDNF in an organotypic culture model of Parkinson’s disease. Brain Res. 2011, 1378, 105–118. [Google Scholar] [CrossRef]
- Jaumotte, J.D.; Wyrostek, S.L.; Zigmond, M.J. Protection of cultured dopamine neurons from MPP(+) requires a combination of neurotrophic factors. Eur. J. Neurosci. 2016, 44, 1691–1699. [Google Scholar] [CrossRef]
- Allen, S.J.; Watson, J.J.; Shoemark, D.K.; Barua, N.U.; Patel, N.K. GDNF, NGF and BDNF as therapeutic options for neurodegeneration. Pharmacol. Ther. 2013, 138, 155–175. [Google Scholar] [CrossRef]
- Khalil, H.; Alomari, M.A.; Khabour, O.F.; Al-Hieshan, A.; Bajwa, J.A. Relationship of circulatory BDNF with cognitive deficits in people with Parkinson’s disease. J. Neurol. Sci. 2016, 362, 217–220. [Google Scholar] [CrossRef]
- Fumagalli, F.; Racagni, G.; Riva, M.A. Shedding light into the role of BDNF in the pharmacotherapy of Parkinson’s disease. Pharm. J. 2006, 6, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; He, X.; Meng, X.; Wu, X.; Tong, H.; Zhang, X.; Qu, S. Regulation of glutamate transporter trafficking by Nedd4-2 in a Parkinson’s disease model. Cell Death Dis. 2017, 8, e2574. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-L.; Liu, Y.; Kang, X.-P.; Dou, C.-Y.; Zhuo, R.-G.; Hunag, S.-Q.; Peng, L.; Wen, L. Ginsenoside Rb1 confers neuroprotection via promotion of glutamate transporters in a mouse model of Parkinson’s disease. Neuropharmacology 2018, 131, 223–237. [Google Scholar] [CrossRef]
- Sveinbjornsdottir, S. The clinical symptoms of Parkinson’s disease. J. Neurochem. 2016, 139 (Suppl. 1), 318–324. [Google Scholar] [CrossRef]
- Rodriguez, M.; Sabate, M.; Rodriguez-Sabate, C.; Morales, I. The role of non-synaptic extracellular glutamate. Brain Res. Bull. 2013, 93, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Dal Ben, M.; Bottin, C.; Zanconati, F.; Tiribelli, C.; Gazzin, S. Evaluation of region selective bilirubin-induced brain damage as a basis for a pharmacological treatment. Sci. Rep. 2017, 7, 41032. [Google Scholar] [CrossRef] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH image to imageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, RESEARCH0034. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | 3 h | 12 h | 24 h | 48 h | 96 h |
|---|---|---|---|---|---|
| Redox imbalance | |||||
| Hmox1 | 1.68 ± 0.40 ** | 1.03 ± 0.16 | 0.64 ± 0.13 | 1.07 ± 0.21 | 1.04 ± 0.46 |
| Srnx1 | 1.21 ± 0.35 | 1.71 ± 0.57 * | 1.48 ± 0.38 ** | 1.69 ± 0.80 * | 1.70 ± 0.39 * |
| Inflammation | |||||
| Tnfa | 1.68 ± 0.54 * | 1.47 ± 0.39 | 1.26 ± 0.19 * | 1.58 ± 0.78 | 0.89 ± 0.29 |
| Cox2 | 1.89 ± 0.55 * | 1.40 ± 0.74 | 2.43 ± 0.69 *** | 0.43 ± 0.58 | 0.31 ± 0.21 |
| Il6 | 0.82 ± 0.44 | 2.10 ± 0.60 * | 5.52 ± 0.81 *** | 0.92 ± 0.68 | 0.88 ± 0.46 |
| Cd68 | 1.35 ± 0.36 | 0.93 ± 0.19 | 0.87 ± 0.15 | 2.16 ± 0.82 | 4.20 ± 1.91 *** |
| Il1b | 1.58 ± 0.51 | 1.07 ± 0.37 | 0.73 ± 0.23 | 0.83 ± 0.45 | 0.39 ± 0.12 * |
| Glutamate neurotoxicity | |||||
| Glu | 1.12 ± 0.2 | 1.34 ± 0.12 | 1.38 ± 0.21 * | 1.53 ± 0.37 ** | 1.52 ± 0.54 ** |
| Neurotrophic growth factor | |||||
| Bdnf | 1.16 ± 0.29 | 0.84 ± 0.48 | 2.01 ± 0.68 *** | 0.19 ± 0.04 ** | 0.17 ± 0.17 ** |
| Sinucleopathy | |||||
| Snca | 0.80 ± 0.21 | 1.38 ± 0.14 | 1.06 ± 0.27 | 1.09 ± 0.48 | 0.90 ± 0.60 |
| Ubiquitination | |||||
| Uchl1 | 0.88 ± 0.17 | 1.08 ± 0.14 | 1.13 ± 0.24 | 0.63 ± 0.19 | 0.46 ± 0.22 ** |
| Vesicular transport | |||||
| Vmat2 | 1.11 ± 0.21 | 1.16 ± 0.56 | 0.46 ± 0.28 | 1.07 ± 0.38 | 0.47 ± 0.11 |
| Apoptosis | |||||
| Anxa5 | 1.20 ± 0.14 | 1.20 ± 0.31 | 1.00 ± 0.23 | 1.57 ± 0.37 * | 1.79 ± 0.58 *** |
| Gene | Accession Number | Forward 5′-3′ | Reverse 3′–5′ |
|---|---|---|---|
| Gapdh | NM_017008.2 | CTCTCTGCTCCTCCCTGTTC | CACCGACCTTCACCATCTTG |
| Tbp | NM_001004198.1 | CAATGACTCCTATGACCCCT | TTTACAGCCAAGATTCACGG |
| Bdnf | NM_012513.4 | GGACATATCCATGACCAGAA | GGCAACAAACCACAACAT |
| Uchl1 | NM_017237.3 | GGAACTGAAGGGACAAGAAG | ATCCATCCTCAAATTCCAGC |
| Vmat2 | NM_013031.1 | AACGTCGCCAAATGTTTAAC | CAATGGATGGTGGGACTAAG |
| Snca | NM_019169.2 | ACCCCTCTTGCATTGAAATT | CATGAACACATCCATGGCTA |
| Hmox1 | NM_012580.2 | GGTGATGGCCTCCTTGTA | ATAGACTGGGTTCTGCTTGT |
| Srxn1 | NM_001047858.3 | AAGGCGGTGACTACTACT | TTGGCAGGAATGGTCTCT |
| Tnfα | NM_012675.2 | CAACTACGATGCTCAGAAACAC | AGACAGCCTGATCCACTCC |
| Il6 | NM_012589.1 | GCCCACCAGGAACGAAAGTC | ATCCTCTGTGAAGTCTCCTCTCC |
| IL1β | NM_031512.2 | AACAAGATAGAAGTCAAGA | ATGGTGAAGTCAACTATG |
| Cox2 | NM_017232.3 | CTTTCAATGTGCAAGACC | TACTGTAGGGTTAATGTCATC |
| CD68 | NM_001031638.1 | ACTTGGCTCTCTCATTCC | GACTGTACTGTGGCTCTG |
| Anxa5 | NM_013132.1 | TAATGACCAAAGCTGTCTCG | TTGATTGACAGCACTTCCAA |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dal Ben, M.; Bongiovanni, R.; Tuniz, S.; Fioriti, E.; Tiribelli, C.; Moretti, R.; Gazzin, S. Earliest Mechanisms of Dopaminergic Neurons Sufferance in a Novel Slow Progressing Ex Vivo Model of Parkinson Disease in Rat Organotypic Cultures of Substantia Nigra. Int. J. Mol. Sci. 2019, 20, 2224. https://doi.org/10.3390/ijms20092224
Dal Ben M, Bongiovanni R, Tuniz S, Fioriti E, Tiribelli C, Moretti R, Gazzin S. Earliest Mechanisms of Dopaminergic Neurons Sufferance in a Novel Slow Progressing Ex Vivo Model of Parkinson Disease in Rat Organotypic Cultures of Substantia Nigra. International Journal of Molecular Sciences. 2019; 20(9):2224. https://doi.org/10.3390/ijms20092224
Chicago/Turabian StyleDal Ben, Matteo, Rosario Bongiovanni, Simone Tuniz, Emanuela Fioriti, Claudio Tiribelli, Rita Moretti, and Silvia Gazzin. 2019. "Earliest Mechanisms of Dopaminergic Neurons Sufferance in a Novel Slow Progressing Ex Vivo Model of Parkinson Disease in Rat Organotypic Cultures of Substantia Nigra" International Journal of Molecular Sciences 20, no. 9: 2224. https://doi.org/10.3390/ijms20092224