Manipulation of Metabolic Pathways and Its Consequences for Anti-Tumor Immunity: A Clinical Perspective


1
Kidney Research Institute, Department of Nephrology, Chang Gung Memorial Hospital, College of Medicine, Chang Gung University, Taoyuan 333, Taiwan
2
Division of Allergy, Asthma, and Rheumatology, Department of Pediatrics, Chang Gung Memorial Hospital, College of Medicine, Chang Gung University, Taoyuan 333, Taiwan
3
Bloomberg~Kimmel Institute for Cancer Immunotherapy, Sidney Kimmel Comprehensive Cancer Center, Department of Oncology, Johns Hopkins University School of Medicine, Baltimore, MD 21287, USA
4
Department of Medical Education, Chang Gung Memorial Hospital at Linkou, Taoyuan 333, Taiwan
*
Authors to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(11), 4030; https://doi.org/10.3390/ijms21114030
Submission received: 13 May 2020
/
Revised: 28 May 2020
/
Accepted: 1 June 2020
/
Published: 4 June 2020
(This article belongs to the Special Issue Cancer Immunotherapy Using Checkpoint Inhibitors: Future Directions 2.0)
Abstract
:In the relatively short history of anti-tumor treatment, numerous medications have been developed against a variety of targets. Intriguingly, although many anti-tumor strategies have failed in their clinical trials, metformin, an anti-diabetic medication, demonstrated anti-tumor effects in observational studies and even showed its synergistic potential with immune checkpoint inhibitors (ICIs) in subsequent clinical studies. Looking back from bedside-to-bench, it may not be surprising that the anti-tumor effect of metformin derives largely from its ability to rewire aberrant metabolic pathways within the tumor microenvironment. As one of the most promising breakthroughs in oncology, ICIs were also found to exert their immune-stimulatory effects at least partly via rewiring metabolic pathways. These findings underscore the importance of correcting metabolic pathways to achieve sufficient anti-tumor immunity. Herein, we start by introducing the tumor microenvironment, and then we review the implications of metabolic syndrome and treatments for targeting metabolic pathways in anti-tumor therapies. We further summarize the close associations of certain aberrant metabolic pathways with impaired anti-tumor immunity and introduce the therapeutic effects of targeting these routes. Lastly, we go through the metabolic effects of ICIs and conclude an overall direction to manipulate metabolic pathways in favor of anti-tumor responses.
1. The Tumor Microenvironment and Immune Checkpoint Inhibitors
Of all diseases, cancer has long been one of the major health concerns in spite of extensive research exploring ways to tackle it. Due to its abnormal growth and deadly metastasis irrespective of limited oxygen and nutrients, the deregulating cellular energetics and the immunoevasion nature of cancer have been identified, which are largely derived from its ability to recruit normal cells that constitutively create the “tumor microenvironment” (TME) [1]. The TME is characterized by a complex interplay between tumor cells and their surrounding neighbors, including stromal cells, extracellular matrix, adipocytes, mesenchymal stem cells, blood vessels, macrophages, T cells, B cells, cytokines, exosomes and metabolites. All the components within the TME contribute to building a 3-dimensional structure with gradients of oxygen tension and availability to nutrients (as shown in Figure 1) [2,3], which favors the development of tumors in multiple aspects such as local progression and distal metastasis [4,5,6,7,8,9]. Importantly, although immune cells are known to protect us from tumors through their immunosurveillance and tumoricidal nature [10,11], multiple factors within the TME may not only hinder their antitumor function but also skew them to construct an immunosuppressive environment in favor of tumor growth.
In the devastating battlefield against tumors, a variety of protumoral factors and immunosuppressive mechanisms have been identified, of which the immunosuppressive cells, exosomes and the co-inhibitory signals play central roles to allow tumor progression. To begin with, the immunomodulatory cells, including but not limited to regulatory T (Treg) cells and M2 macrophages, accumulate in the TME and diminish T cell anti-tumor immune responses [12,13]. Treg cells are famous for their immunosuppressive effects on not only aberrant immune responses against self-antigens but also anti-tumor immune responses, in both laboratory and clinical studies [14,15,16]. Treg cells are believed to modulate immune responses through expressing immunosuppressive cytokines (including transforming growth factor (TGF)-β, interleukin (IL)-10, IL-35), immune checkpoints (such as cytotoxic T-lymphocyte-associated antigen (CTLA)-4; programmed death (PD)-1) and other co-inhibitory receptors, as extensively reviewed by previous studies [17,18]. Furthermore, Treg cells are also capable of inducing tolerogenic dendritic cells (DCs) that are linked to T cell exhaustion, and can release cytotoxic agents such as perforin and granzyme [19,20,21,22,23]. Therefore, the aberrant accumulation of Treg cells in TME impairs anti-tumor immunity through various mechanisms [24,25], facilitating tumor growth and progression. Besides Treg cells, the unwanted immunosuppression within the TME can also be affected by tumor-associated macrophages (TAMs) that are majorly driven by cytokines such as IL-4 or IL-13, which have also earned their “M2-like” naming [26,27,28]. Orchestrated by signals from tumor cells, T cells as well as stroma [29,30], TAMs play a protumoral role in the TME by promoting tumor metastasis through promoting angiogenesis as well as extracellular matrix remodeling [31,32]. Moreover, TAMs also exert their profound immunosuppressive effects through expressing a variety of inhibitory ligands and cytokines, including PD-L1, PD-L2, B7-1, B7-2, HLA-G, HLA-E, IL-10 as well as TGF-β, as clearly reviewed by previous studies [33,34]. Besides immunosuppressive cells, previous studies further reveal the vital roles of exosomes and co-inhibitory molecules to promote tumor growth and metastasis in many different ways.
The aberrant accumulation of immunosuppressive cells within the TME is thought to be affected by both exosomes and co-inhibitory signals derived from the tumor cells. Exosomes are extracellular vesicles with bi-layered membrane that range between 30-100 nm in diameter [35], while patients with cancer, especially of the ones with poor prognosis, are often found with higher numbers of them [36]. Tumor-derived exosomes have been detected in many different types of cancers, and they can not only impair anti-tumor immunity within the TME but also educate bone marrow-derived progenitor cells to facilitate distant metastasis [37,38,39,40]. On the other hand, the expression of co-inhibitory molecules such as PD-L1 have been observed on various kinds of cancer cells [41], which correlates with poor clinical outcomes of many patients with cancer [42,43]. Importantly, since the co-inhibitory checkpoint molecules expressed by both tumors and immunosuppressive cells within the TME dampens anti-tumor immunity of T cells [44,45,46,47], immune checkpoint inhibitors (ICIs) such as the antibodies directed against CTLA-4, PD-1 and PD-L1, are developed into encouraging treatments against various tumors, most notably melanoma and non-small cell lung cancer [48,49,50,51]. For instance, a pooled meta-analysis assessing long-term survival of 1861 advanced melanoma patients estimated a 3-year survival rate of 22% for patients receiving anti-CTLA4 ipilimumab [52], which evidently outperformed other chemotherapy such as dacarbazine, where the 3-year survival rates were only around 12% [53]. However, despite the game-changing efficacies of ICIs against tumors in clinical scenarios, they still face improvable downsides, including the suboptimal long-term response rates due to both innate and acquired resistance [54], as well as the lack of a reliable predictive biomarker [55]. Therefore, a variety of combinatory therapeutic strategies, along with the discovery of potential biomarkers, have been tested out to overcome the limitations, and targeting the metabolic pathways within the TME has appeared as one of the emerging candidates that have synergistic potential.
2. The Implications of Metabolic Syndrome for Targeting Metabolic Pathways within the Tumor Microenvironment
When it comes to the phrase “metabolic”, metabolic syndrome pops up in many physicians’ minds due to its extremely high prevalence as well as its modifiable nature [56,57,58]. As a cluster of unfavorable conditions including increased blood pressure, blood sugar, cholesterol levels, and body fat around the waist, the poor control of metabolic syndrome is widely believed to result in metabolic diseases such as type 2 diabetes mellitus [59,60,61], which is characterized by hyperglycemia and hyperinsulinemia due to increased insulin resistance. Although these pathological conditions are believed to lead to further complications such as stroke, heart attack, diabetic foot and diabetic nephropathy, accumulating epidemiological evidence also confirms their close relationships with not only worse prognosis in tumor patients but also an increased risk for tumor [62,63,64,65,66,67,68]. For instance, a meta-analysis study sums up the epidemiological evidence, indicating that the incidences of various cancers, including pancreas, liver, breast, colorectal, urinary tract and female reproductive organs, are increased in diabetic patients [69]. Furthermore, a recent study has estimated that of the documented global tumor cases in 2012, 2.0% of them were attributable to diabetes mellitus alone, whereas 5.6% of them were attributable to the combined effects of both diabetes and high BMI [64]. Although systemic manifestations such as hyperglycemia, insulin resistance, hyperinsulinemia and chronic inflammation have provided clues to the origin of these associations [70,71,72,73,74,75,76,77], it is difficult to delineate the underlying mechanisms based on macro-level perspectives without knowing how each pathological molecule affects the pathogenesis. Therefore, physicians must look into the subcellular and molecular course of events, such as the metabolic pathways.
Metabolic pathways are the consecutive series of biochemical reactions that take place in a single cell, enabling a variety of cellular functions. Besides the discussed factors that favor tumor growth within the TME, it is also indispensable to keep an eye on the related metabolic pathways, especially of the one involving the insulin-like growth factor (IGF) system, to elucidate the underlying connection between metabolic syndrome and tumor [78]. The IGF system consists of three molecules that induce their cellular responses mainly through IGF-1 receptor (IGF-1R), namely insulin, IGF-1 and IGF-2 [79]. IGF-1 is a 70-aminoacid polypeptide hormone mainly produced by the liver [80], and it shares structural homology with IGF-2 and proinsulin [81]. IGF-1 can either bind to its putative receptor (IGF-1R) or the insulin receptor, while there is also a hybrid form of receptor which both insulin and IGF-1 can ligate to [82,83]. After binding, these receptors elicit downstream tyrosine kinase activity and activate the Ras-mitogen-activated protein kinase and phosphatidylinositol 3-kinase (PI3K)/Akt pathways [84,85]. Whilst the Ras-mitogen-activated protein kinase mediates the mitogenic effect, the PI3K/Akt pathway activates enzymes involved in gluconeogenesis, glucose uptake, protein synthesis and lipogenesis, reprogramming the metabolic pathways [79,86]. In tumor cells, the IGF-1 signaling drives neoplastic behaviors [87,88,89], as well as protects tumors from cytotoxic treatment likely through promoting the activity of efflux pumps while upregulating double-strand break repair [90,91,92]. The clinical relevance of these in vitro findings was confirmed by, for example, an immune-histochemical analysis of pancreatic cancer biopsies that identified activated insulin/IGF receptors expressed in 72% of the patients, which positively correlates with the infiltration of TAMs that secrete IGF-1 and IGF-2 along with tumor stromal myofibroblasts, supporting the chemo-resistance of the tumors [93]. Furthermore, the insulin receptor was found to be overexpressed by the angiogenic vasculature in human tumors, facilitating tumor angiogenic activity and correlates to shorter survival [94]. In addition, hyperinsulinemia also promotes carcinogenesis indirectly by reducing the hepatic production of IGF binding proteins [95,96,97], thereby elevating the circulating levels of bioactive IGF-I and, therefore, further fuels the growth of neoplastic cells [78,98]. On the other hand, although the endocrine-immune axis of the IGF system is much more complicated [99,100], evidence has suggested its overall immunosuppressive effects by increasing the infiltration of Treg cells in affected tissues [101,102,103], enhancing the expression of immunosuppressive cytokines as well as inducing the M2 phenotype of macrophages that secretes anti-inflammatory cytokines [104,105,106,107]. In short, the excessive insulin in patients with metabolic syndromes facilitates these metabolic pathways in the TME, which potentiates tumor growth and compromises antitumor activity. After reviewing the basic findings in metabolic pathways, it is, therefore, unsurprising for physicians to observe that tumor risk is higher [108], as well as that the prognosis of some tumors is worse in patients with hyperinsulinemia, shedding light on the therapeutic potentials of targeting these metabolic pathways [72,109].
Before developing targeted therapy against the aberrant IGF system within the TME, clinical observations had revealed the close relationship between certain anti-diabetic strategies and their protective effects against cancer. To begin with, losing weight is known to improve not only the outcome of type 2 diabetes through enhancing overall glycemic and metabolic control [110,111], but also provide anti-tumor benefits in clinical scenarios [112,113,114]. This approach increases serum IGFBP-2 level and decreases the number of infiltrating macrophages in both adipose tissue and tumors of colon and liver [115,116,117], likely improving the prognosis through reducing the metabolic signals originated from the IGF system. Second, as the long been preferred first-line oral anti-diabetic agent to manage type 2 diabetes [118], Metformin exerts its anti-diabetic effects through decreasing hyperinsulinemia and insulin resistance [119,120], but it is also known of its anti-tumor activity. In vitro studies of Metformin showed that it inhibits tumor cell proliferation through partial cell cycle arrest [121,122,123] and activates 5′ adenosine monophosphate-activated protein-activated protein kinase in tumor cells, leading to growth inhibition by inhibiting protein synthesis [124]. Furthermore, in vivo studies have pointed out that metformin has more anti-tumor activities in mice receiving a high-energy diet, associated with hyperinsulinemia and accelerated tumor growth, than it does in mice receiving a balanced diet [125], indicating that the insulin-lowering effect of metformin likely contributes to its anti-tumor activity [126,127]. Besides, clinical evidence also suggests that elevated insulin and IGF-1 levels favor the progression of a variety of cancers [128,129]. Therefore, it is not surprising that a substantial number of observational human studies suggest that treatment with metformin reduces cancer risk as well as cancer mortality [130,131,132,133,134,135], which is in clear contrast to the increased risk for cancer or cancer death associated with the use of exogenous insulin, insulin analog glargine or sulfonylureas [136,137,138]. For instance, a systemic review and meta-analysis have noted a 31% reduction in overall summary relative risk for cancer in patients taking metformin compared with other antidiabetic medications and a trend to a dose–response association was also found [139]. In contrary, it was reported that insulin glargine, which would be ultimately required by many diabetic patients [140], has elevated mitogenic potency [89,141,142], supporting the aforementioned clinical findings as well as the importance of this metabolic pathway in oncology. Although confounding by indication from observational studies may affect the interpretation of results, experimental studies have additionally supported these findings by revealing the metformin-induced energy stress on tumor cells that reduce their mitochondrial ATP output, turning them into a more glycolysis-dependent state [143,144], especially in tumors lacking LKB1 or p53 [126,145]. In short, it is evident that the aberrant IGF system contributes, at least partly, to the tumor development and growth, so agents that target this metabolic axis have been tested out.
Several inhibitors of the IGF system have been developed as anti-tumor treatments, including small molecules that inhibit PI3K or its downstream signal Akt [146,147]. In addition, medications such as sorafenib have also demonstrated ability to reduce the expression of IGF-1 by M2 macrophages as well as the following IGF-1-driven hepatocellular carcinoma growth in a preclinical study [148]. Although the rationale of targeting this pathway as well as its preclinical studies seem promising, the single-agent strategy has yet to find great success in clinical trials [149,150,151]. Besides the undesired inhibition of insulin action in the liver, muscle and fat that results in metabolic toxicity and likely precludes their use at clinically effective doses [152], monotherapy against the IGF system may also downregulate anti-tumor immunity. For instance, it was demonstrated that the silencing of the insulin receptor reduced glucose transport and glycolysis in activated CD4+ T cells, resulting in enhanced apoptosis [153]. Furthermore, knockdown of the insulin receptor also reduces cytotoxic responses and proliferation rates of CD8+ T cells, while leaving the immunosuppressive Treg cells unaffected [153]. Taking into account the downsides of targeting this pathway alone, various combinatory therapies have been investigated. For instance, in vivo studies have noted that the inhibition of IGF-1/IGF-2 sensitized pancreatic tumors to gemcitabine [93], whereas combined IGF-1R and CSF-1R inhibition significantly improved survival outcomes in mice with glioblastoma multiforme [154]. Furthermore, since downregulating IGF-1 or IGF-1R has been found to enhance the immunogenicity of GBM models in rats and a small clinical study in astrocytoma [155,156,157], possibly partly contributed by inducing tumor cell death to release tumor-specific antigens [158], further investigation of the anti-tumor efficacy of combining ICIs with anti-IGF strategies is also warranted.
Currently, although there may be inadequate evidence to evaluate the effect of combining ICIs with anti-IGF treatment against tumors, the combination of ICIs with metformin did show some encouraging results. A retrospective cohort study of patients with metastatic malignant melanoma revealed favorable treatment-related outcomes in terms of objective response rate, disease control rate, overall survival and progression-free survival in patients who received metformin in combination with ICIs, as compared to patients receiving ICIs alone [159]. Another retrospective descriptive analysis of the randomized phase III OAK trial for the treatment of advanced or metastatic previously treated non-small cell lung cancer also demonstrated a synergistic benefit of metformin treatment in addition to the anti-PD-L1 antibody atezolizumab in the overall response rate [160]. The observed anti-tumor potential of metformin may not only derive from downregulating the IGF system by decreasing hyperinsulinemia but also contributed by regulating immunometabolisms [161]. By blocking mTOR signaling and restoring mitochondrial fatty acid oxidation, metformin facilitates the shift of T cell metabolism from a glucose-dependent anabolic state to a catabolic state of metabolism, which resembles memory T-cell [162,163]. Therefore, it may not be surprising that a clinical trial of head and neck squamous cell carcinoma showed that metformin use was linked to a 66.5% increase in CD8+ effector T cells and a 41.4% decrease in FoxP3+ Treg cells within the TME [164]. Although metformin is likely to work synergistically with ICIs by modulating metabolic pathways of tumor cells and immune cells, the observed benefits may also contribute to its ability to not only inhibit the tolerogenic M2-TAMs [165], plausibly via modifying their lipid metabolism and kynurenine metabolic pathway [166,167], but also enhance PD-L1 degradation [168]. In light of the possible therapeutic mechanisms, it is also worth noting that these combinatory treatments may identify suitable patient populations and further optimize treatment outcomes by developing predictive biomarkers (e.g., hyperinsulinemia) based on the information.
3. The Metabolic Pathways in the Tumor Microenvironment: Glucose and Lipid
Besides the aforementioned metformin and IGF system, many other developing strategies aim to direct metabolic pathways of immune cells and/or tumor cells to overcome the immunosuppressive environment within the TME. As introduced in the previous sections, despite a variety of immune cells being found within the TME, such as DCs, macrophages, T cells, as well as B cells, plasma cells and so on, they either educate each other through anti-inflammatory signals to construct an immunosuppressive condition or face a hostile environment consisting of nutrient scarcity, metabolite abundance and hypoxia [169]. Cytotoxic CD8+ T lymphocytes are known to be the main antitumor effector cells [170,171] which initiate their tumoricidal function after priming by antigen-presenting cells (APCs) such as macrophages and DCs [172,173]. However, although TAMs are considered as the most abundant APCs within the TME, they often have poor antigen-presenting capacity [172,174]. On the other hand, the priming process can neither rely on DCs, since both intratumoral and systemic DCs are found to be dysfunctional and/or reduced [175,176,177]. The dysfunctional, or even skewed, anti-tumor activities were considerably affected by the altered metabolic pathways, which are turning out to be one of the most promising anti-tumor targets.
In fact, cancers favor glycolysis for energy even in the presence of sufficient oxygen, known as the Warburg effect, which forms the basis for diagnostic tools like 18F-fluorodeoxyglucose-positron emission tomography [178]. The net gain of anaerobic oxidation of 1 mole of glucose is 2 moles of ATP, which is far less than the following oxidative phosphorylation that generates 36 additional moles of ATP, this means a much higher amount of glucose consumed for energy maintenance under anaerobic conditions. Since glycolysis is also required by effector T cells [179,180,181,182], as well as activated macrophages and DCs [183,184], the competitive TME impairs T cells by limiting their mTOR activity as well as glycolytic capacity [185,186], while interrupting the metabolic programs and promoting the development of tolerogenic DCs and M2-like macrophages [187,188,189]. Furthermore, besides reprograming Th17 cells into Treg cells [180], limited glycolysis also inhibits the expression of interferon-γ by Th1 cells [190,191] and hinders the anti-tumor effector functions of Th1 CD4+ T cells while upregulating TGF-β production [186]. Interestingly, it was reported that inhibited glycolysis during CD8+ T cell differentiation skews effector T cell generation towards a more central memory phenotype [192,193]. On the other hand, the TCA cycle and oxidative phosphorylation are associated with a more anti-inflammatory phenotype [194,195,196,197] and are prominent in memory CD8+ T cells [198]. In contrast to glycolysis, generating energy through fatty acid oxidation (FAO) has been observed in many non-inflammatory immune cells with increased cellular lifespans, including M2 macrophages, Treg cells and memory T cells [199]. FAO promotes the generation of Treg cells while inhibiting effector T cell polarization in favor of tumor growth [179]. However, FAO also plays a key role in the generation and maintenance of memory CD8+ T cells, which is being acknowledged as the main contributor of long-lived immunity and activated DCs against tumors [200,201]. Memory CD8+ T cells appear to rely on FAO to have timely responses to antigen stimulation [202], while treatment like IL-15 promotes their FAO and results in increased cell survival [203]. In addition, since targeting lipid synthesis in tumor cells inhibits tumor progression [204,205,206], whether it also affects the immune system has been evaluated. It was found that increased fatty acid synthesis (FAS) is necessary for DC activation and their subsequent stimulation of CD8+ T cells [207]. FAS is also important for both T cells and B cells, especially for cell proliferation after activation [208,209]. Intriguingly, T cell-specific deletion of acetyl-CoA carboxylase 1 (ACC1), the rate-limiting enzyme in FAS, was found to reduce blasting efficacy along with a lower accumulation of antigen-specific CD8+ T cells, which could be reversed by supplementing them with exogenous fatty acids [210]. However, neither pharmacological nor genetic inhibition of ACC1 in CD4+ T cell subsets seems to affect Treg cell generation and function [211]. Due to the multifaceted influences of targeting metabolic pathways for anti-tumor therapy, there could plausibly be inefficacies in their monotherapy, which are expected to be overcome by combinatory treatments.
Since the TME is depleted of glucose [212], it inhibits CD4+ effector T cells and promotes Treg cells, generating an immunosuppressive environment. Therefore, several strategies have been under investigation to rewire the aberrant metabolic reprogramming. Enhancing memory-associated metabolism, including oxidative phosphorylation and FAO, by small-molecule inhibitors which can enforce mitochondrial fusion or inhibit mitochondrial fission, generated CD8+ T cells and controlled tumor growth in vivo [213]. Furthermore, several studies have demonstrated that the co-stimulatory receptor 4-1BB is key to maintaining mitochondrial health and biogenesis for robust anti-tumor immunity [214,215]. Similarly, metabolic interventions can also be directed against tumor cells, as inhibiting tumor glycolysis also improves T cell-mediated anti-tumor effects both in vitro and in vivo [216], while another study has shown that the efficacy of ICI therapy was promoted by the inhibitory effect of metformin on oxygen consumption of tumor cells, thereby reducing intra-tumoral hypoxia and potentiating anti-tumor immunity [217]. In short, treatments that open a door to rewire the TME-associated aberrant metabolic landscape concerning insufficient glycolysis accompanied by different degrees of hypoxia hold the potential to aid in future anti-tumor strategies.
When it comes to improving treatments against tumors by manipulating metabolisms of fatty acids, targeting FAO, preferentially utilized by non-inflammatory and tolerogenic immune cells and FAS, resembled inflammatory responses, have gradually discovered encouraging findings [199]. It is known that regulating cholesterol metabolism in CD8+ T cells is important to maintain a strong TCR clustering during activation [218]. As a result, Avasimibe, an inhibitor of the cholesterol esterification enzyme acetyl-CoA acetyltransferase (ACAT) 1, was developed, demonstrating enhanced proliferation as well as effector function of CD8+ T cells [218]. In addition, TVB-2640, an inhibitor of FAS, had entered early clinical trials in light of the critical role that FAS plays in early T cell activation and clonal expansion, demonstrating prolonged stable disease across multiple tumor types [219]. Taken together, these studies point out the anti-tumor potential of targeting lipid metabolisms, which may work synergistically with the current state of ICIs.
4. The Metabolic Pathways in the Tumor Microenvironment: Amino Acids and Metabolic Intermediates
Effector T cells consume large amounts of glucose, the carbons of which are largely secreted as lactate [220], whereas amino acids serve as building blocks to support protein and nucleotide synthesis, which are especially required upon T cell activation [221]. To begin with, the important role for glutamine to fuel the TCA cycle and generate metabolic intermediates for anabolic proliferation has been well-documented in both cancer and immune cells [221,222]. It was noted that there is a marked increase in glutamine usage upon both T cell and B cell activation in order to respond to antigen stimulation [222,223,224]. Glutamine metabolism is also suggested to regulate the balance between effector T cells and Treg cells, because the impaired uptake of neutral amino acids caused by the genetic loss of alanine serine cysteine transporter 2 (ASCT2) in T cells results in hindered generation and function of Th1 and Th17 cells, while not affecting Treg cell generation [225,226]. Furthermore, both the reduction of glutamine levels in culture media and ASCT2 loss reduced mTORC1 activity, which coincided with the effector T cell defects [225]. Therefore, although glutamine antagonists such as 6-diazo-5-oxo-L-norleucine (DON) have the ability to inhibit tumor growth in clinical scenarios [227], likely thereby improving overall anti-tumor immunity, whether they would also hinder anti-tumor immunity metabolically remains controversial. DON is a glutamine analogue that can bind to the catalytic centers of various glutamine-utilizing enzymes such as GLS1 (glutaminase1), achieving glutamine blockade of many metabolic pathways [228,229]. Recently, the anti-tumor effects of glutamine blockade with the prodrug of DON, JHU-083, has been tested out in tumor-bearing mice [230]. It was found that glutamine blockade not only decreased hypoxia, acidosis and nutrient depletion within the TME by suppressing oxidative and glycolytic metabolism of cancer cells, but also markedly upregulated oxidative metabolism of CD8+ effector T cells, adopting a long-lived and highly activated phenotype. These studies proved to be highly instructive in terms of better understanding the differences in T cell and cancer metabolism as well as novel concepts concerning the metabolism of anti-tumor specific T cells. Indeed, glutamine helps to support massive clonal expansion of T cells and blocking glutamine pathways can limit the magnitude of clonal expansion. However, recall that after peak expansion there is significant contraction characterized by cell death. It was found that while inhibiting glutamine metabolism does limit peak expansion, it also reprograms the T cell metabolically such that they are more robust and do not undergo massive contraction. In the tumor setting, blocking glutamine metabolism markedly inhibits tumor growth while simultaneously reprogramming anti-tumor T cells to be more robust memory and effector cells. Additionally, glutamine metabolism also plays a critical role in M2 macrophage polarization by fueling the TCA cycle [194], where inhibition of glutamine synthetase skews M2 macrophages to an M1-like phenotype and prevents tumor metastasis [231]. Likewise, it was found that inhibition of glutamine metabolism employing the DON prodrug JHU-083 can inhibit MDSC generation, inhibit metastasis and promote anti-tumor immunity [232]. As a result, multiple clinical trials are being carried out to evaluate the safety and efficacy of glutaminase inhibitors in treating different tumors [233,234,235], and a combinatory approach for targeting this metabolic pathway is expected to lead to more promising results.
In addition, there has been much focus on the immune regulatory role of tryptophan metabolism particularly on indoleamine-2, 3-dioxygenase (IDO). The proliferation of T cells requires tryptophan [236], whereas driving IDO expression and subsequent tryptophan catabolism in APCs blunts T cell stimulation [237]. Indeed, tryptophan insufficiency can induce accumulation of charged tRNAs, while activating the unfolded protein response protein GCN2 [238]. Importantly, many other aspects of tryptophan metabolism, especially metabolites generated from tryptophan catabolism such as kynurenine, can modulate immune cell function through activation of the aryl hydrocarbon receptor [239]. IDO expression is generally seen in tumor cells, surrounding stromal cells and even TAM [240,241,242], while increased IDO expression correlates with a poor prognosis in certain cancers [243,244], likely through impairing the anti-tumor effects of T cells [245,246]. Although inhibition of IDO in murine tumor models stimulated the T cell-mediated anti-tumor responses [247,248], a recent phase III study of the direct IDO inhibitor, epacadostat, failed to further increase the progression-free survival in combination with anti-PD-1 therapy over anti-PD-1 monotherapy [249]. Therefore, alternative methods of inhibiting this pathway are being pursued, and, hopefully, may yield further clinical implications upon combining with anti-tumor agents.
Serving as a precursor for the biosynthesis of proteins as well as creatine, arginine is also required for effective immune responses. It was shown that exogenous l-arginine leads to improved T cell fitness as well as enhanced generation of central memory cells [250]. Arginine has also been noted to regulate the T cell receptor [251], as well as to support the proliferation of human T cells [252]. In addition, an adequate supply of arginine is also critical for M1 macrophages to generate nitric oxide for anti-microbial and cytotoxic functions [253]. However, the arginase expression in macrophages has been reported to limit the inflammatory response of effector T cells [254], possibly through suppressing mTORC1 activity due to arginine-depletion that has also been observed in glutamine deficiency [255,256]. This competition over arginine has been observed in various human cancers and mouse cancer models, where TAMs are identified to secrete arginase into the TME [257,258], metabolizing l-arginine and inhibiting T cells [259]. Based on this rationale, CB-1158, a potent inhibitor of arginase, has been under investigation as a strategy to shift the TME toward a pro-inflammatory environment [260]. So far this approach has shown good tolerance along with on-target inhibition [261], and even synergy with ICIs such as pembrolizumab [262].
Excessive amounts of the metabolites lactate and adenosine found in the TME serve to inhibit anti-tumor immunity. Many cancers rely on aerobic glycolysis, which produces a large amount of lactate and acidifies the TME, helping tumor growth while evading the anti-tumor responses [216,263,264]. Lactate can suppress immune responses against tumor by driving M2-like macrophage polarization [189,265], inhibiting NK cell activity [266], conversely favoring Treg cells [267], as well as blocking proliferation and functions of T cells [268,269,270]. As a result, there has been much interest in targeting l-lactate dehydrogenase (LDH) A chain, which catalyzes the formation of lactic acid [271]. Nevertheless, it is important to point out that effector T cells also rely on the activity of LDH for effective proliferative and functional responses [272,273]. Again, monotherapy that blocks LDH may, therefore, have unwanted effects in addition to the effector arm of the anti-tumor response [274]. Besides lactate, the concentration of adenosine is also markedly increased in hypoxic tissues and the TME [275], supporting angiogenesis and profoundly suppressing immunity [276,277]. Both CD39 and CD73 are cell surface molecules with critical roles in controlling the production of adenosine [278] and their upregulations in tumors are linked to poor prognosis [279,280,281]. Furthermore, Treg cells can also express CD39, causing immunosuppression as well as the abrogation of NK cell function in the TME [282,283]. Besides, adenosine also inhibits T cells from expressing the IL-2 receptor and TCR-stimulated proliferation [284], while upregulating the expression of immune-checkpoint receptors (including PD-1 and CTLA-4) [285]. The generally immunosuppressive effects discovered of this pathway paved the way for many pre-clinical studies, showing that targeting the adenosine-related pathways had synergistic anti-tumor effects along with ICIs [286,287,288]. An early clinical trial revealed that the use of CPI-144, an adenosine A2a receptor inhibitor, was associated with increasing disease control over refractory renal cell carcinoma and also acts synergistically with ICI [289], and there are currently a number of ongoing clinical trials [290].
To sum up, amino acids, along with lactate, generally have diverse immunomodulatory effects, which possibly require cell-specific manipulations or combinatory strategies to specifically rewire the metabolic pathways needed to maximize the efficacy of anti-tumor activity. On the other hand, targeting metabolites such as adenosine have yielded promising synergistic effects with ICIs so far, but further investigation is warranted.
5. The Effects of Exosomes and Immune Checkpoint Inhibitors on Metabolic Pathways
Since the immunosuppressive nature of the TME may largely be affected by both exosomes and co-inhibitory molecules, as discussed previously, looking into their effects on metabolic pathways is important for developing novel therapeutic strategies. To begin with, the acidified environment built up by lactate facilitates both the release and uptake of exosomes [291]. It was reported that gastric cancer-derived exosomes could transmit autocrine signals that promote tumor proliferation via increasing the phosphorylation of Akt [292]. Furthermore, as one of the major cell types within the TME, cancer-associated fibroblasts have been shown to produce exosomes that inhibit oxidative phosphorylation and increase glycolysis as well as glutamine-dependent reductive carboxylation in tumor cells [293]. In addition, these exosomes could also fuel nutrient-deprived tumor cells with TCA intermediates and glutamine [294]. Similarly, exosomes secreted by adipocytes have also been found to transmit proteins implicated in FAO that promote tumor migration and invasion [295]. Despite these intriguing findings, whether they could be further developed into successful treatment in clinical scenarios requires further research.
Given the therapeutic efficacy of ICIs in many cancers, it may not be surprising to find that they also influence metabolic programming as well as the nutrient competition between tumor cells and T cells. Interaction of PD-1 with its ligands promotes FAO [296], but prevents metabolic reprogramming including upregulation of aerobic glycolysis as well as glutaminolysis, in T cells through inhibiting the PI3K–Akt–mTOR pathway [297]. Interestingly, PD-L1 has also been found to affect the metabolism of tumor cells by stimulating aerobic glycolysis through activating the PI3K–Akt–mTOR pathway [185]. As a result, by inhibiting the PD-1–PD-L1 axis, a profound anti-tumor effect could be expected by restoring the metabolic fitness of T cells while downregulating aerobic glycolysis in tumor cells, thereby also increasing the availability of glucose to the T cells [216]. In addition to the PD-1–PD-L1 axis, the CD28-CTLA-4 axis also affects metabolic pathways. It is known that reducing Akt phosphorylation and activation contributes to the inhibitory effect of CTLA4 signaling on CD28-mediated co-stimulation [298], which possibly impairs the increased glucose metabolism and mitochondrial remodeling following T cell activation. However, unlike PD-1, CTLA-4 signaling does not seem to promote FAO [296]. To sum up, ICIs elicit profound anti-tumor activities at least partly affected by their ability to modify metabolic pathways and win the tug-of-war over nutrients in TME, suggesting the importance to incorporate thoughts on metabolic pathways when designing anti-tumor strategies. It is vital to keep in mind, however, that not only do different immune cells depend on different metabolic pathways (as shown in Table 1) but also there are likely to be numerous mechanisms through which each metabolic pathway acts to modulate the immune system. Future work on further elucidating the metabolic effects of various anti-tumor agents on both immune and tumor cells could yield new insights to treat various cancers.
6. Conclusions
In the history of oncology, there have already been numerous strategies against cancers proposed, but only a small proportion of them have proved to be effective in clinical trials. Intriguingly, anti-tumor effects were found not only in the successful development of ICIs but also in various drugs which target metabolism. Looking back from bedside-to-bench, the metabolic pathways within the TME appear to be increasingly important in oncology. There is a strong rationale supporting the combination of metabolic interventions with many other developed therapies against cancer, and a variety of ongoing clinical trials are being carried out [299]. Targeting the metabolic pathways is undoubtedly important in breaching the immunosuppression imposed by the TME, and potentially even directly improving anti-tumor immunity by targeting suppressive cells like Tregs and MDSC, enhancing the generation of memory cells and effector functions.
Funding
This review article was supported by grants from the Chang Gung Memorial Hospital (CORPG3F0131-3, CORPG5H0091).
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| TME | tumor microenvironment |
| Treg | regulatory T |
| TGF | transforming growth factor |
| IL | interleukin |
| CTLA-4 | cytotoxic T-lymphocyte-associated antigen 4 |
| PD-1 | programmed death 1 |
| DC | dendritic cell |
| TAM | tumor-associated macrophage |
| ICI | immune checkpoint inhibitor |
| IGF | insulin-like growth factor |
| PI3K | phosphatidylinositol 3-kinase |
| APC | antigen-presenting cell |
| FAO | fatty acid oxidation |
| FAS | fatty acid synthesis |
| ACC1 | acetyl-CoA carboxylase 1 |
| ACAT | acetyl-CoA acetyltransferase |
| ASCT | alanine serine cysteine transporter |
| DON | 6-diazo-5-oxo-L-norleucine |
| IDO | indoleamine-2,3-dioxygenase |
| LDH | lactate dehydrogenase |
| OXPHOS | oxidative phosphorylation |
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Ertl, H.C. Starved and asphyxiated: How can CD8+ T cells within a tumor microenvironment prevent tumor progression. Front. Immunol. 2016, 7, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilkes, D.M.; Semenza, G.L.; Wirtz, D. Hypoxia and the extracellular matrix: Drivers of tumour metastasis. Nat. Rev. Cancer 2014, 14, 430–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meseure, D.; Alsibai, K.D.; Nicolas, A. Pivotal role of pervasive neoplastic and stromal cells reprogramming in circulating tumor cells dissemination and metastatic colonization. Cancer Microenviron. 2014, 7, 95–115. [Google Scholar] [CrossRef] [PubMed]
- Alkasalias, T.; Moyano-Galceran, L.; Arsenian-Henriksson, M.; Lehti, K. Fibroblasts in the tumor microenvironment: Shield or spear? Int. J. Mol. Sci. 2018, 19, 1532. [Google Scholar] [CrossRef] [Green Version]
- Poltavets, V.; Kochetkova, M.; Pitson, S.M.; Samuel, M.S. The role of the extracellular matrix and its molecular and cellular regulators in cancer cell plasticity. Front. Oncol. 2018, 8, 431. [Google Scholar] [CrossRef] [Green Version]
- Almendros, I.; Gileles-Hillel, A.; Khalyfa, A.; Wang, Y.; Zhang, S.X.; Carreras, A.; Farré, R.; Gozal, D. Adipose tissue macrophage polarization by intermittent hypoxia in a mouse model of OSA: Effect of tumor microenvironment. Cancer Lett. 2015, 361, 233–239. [Google Scholar] [CrossRef]
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K. Role of tumor microenvironment in tumorigenesis. J. Cancer 2017, 8, 761. [Google Scholar] [CrossRef]
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; Almeida, D.; Koller, A.; Hajjar, K.A.; Stainier, D.Y. The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol. 2013, 15, 807–817. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Pagès, F.; Sautes-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Nishikawa, H.; Sakaguchi, S. Regulatory T cells in cancer immunotherapy. Curr. Opin. Immunol. 2014, 27, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T cells and immune tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- deLeeuw, R.J.; Kost, S.E.; Kakal, J.A.; Nelson, B.H. The prognostic value of FoxP3+ tumor-infiltrating lymphocytes in cancer: A critical review of the literature. Clin. Cancer Res. 2012, 18, 3022–3029. [Google Scholar] [CrossRef] [Green Version]
- Shevach, E.M. Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity 2009, 30, 636–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, A.; Oberle, N.; Krammer, P.H. Molecular mechanisms of treg-mediated T cell suppression. Front. Immunol. 2012, 3, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelhardt, J.J.; Boldajipour, B.; Beemiller, P.; Pandurangi, P.; Sorensen, C.; Werb, Z.; Egeblad, M.; Krummel, M.F. Marginating dendritic cells of the tumor microenvironment cross-present tumor antigens and stably engage tumor-specific T cells. Cancer Cell 2012, 21, 402–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, X.; Cai, S.F.; Fehniger, T.A.; Song, J.; Collins, L.I.; Piwnica-Worms, D.R.; Ley, T.J. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity 2007, 27, 635–646. [Google Scholar] [CrossRef] [Green Version]
- Veldhoen, M.; Moncrieffe, H.; Hocking, R.J.; Atkins, C.J.; Stockinger, B. Modulation of dendritic cell function by naive and regulatory CD4+ T cells. J. Immunol. 2006, 176, 6202–6210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grossman, W.J.; Verbsky, J.W.; Barchet, W.; Colonna, M.; Atkinson, J.P.; Ley, T.J. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity 2004, 21, 589–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Z.; Garg, S.K.; Banerjee, R. Regulatory T cells interfere with glutathione metabolism in dendritic cells and T cells. J. Biol. Chem. 2010, 285, 41525–41532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vignali, D.A.; Collison, L.W.; Workman, C.J. How regulatory T cells work. Nat. Rev. Immunol. 2008, 8, 523–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Franco, F.; Ho, P.-C. Metabolic regulation of Tregs in cancer: Opportunities for immunotherapy. Trends Cancer 2017, 3, 583–592. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Barreto, J.B.; Andreu, P.; Vasquez, L.; Tawfik, D.; Kolhatkar, N.; Coussens, L.M. CD4+ T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell 2009, 16, 91–102. [Google Scholar] [CrossRef] [Green Version]
- Shiao, S.L.; Ruffell, B.; DeNardo, D.G.; Faddegon, B.A.; Park, C.C.; Coussens, L.M. TH2-polarized CD4+ T cells and macrophages limit efficacy of radiotherapy. Cancer Immunol. Res. 2015, 3, 518–525. [Google Scholar] [CrossRef] [Green Version]
- De Monte, L.; Wörmann, S.; Brunetto, E.; Heltai, S.; Magliacane, G.; Reni, M.; Paganoni, A.M.; Recalde, H.; Mondino, A.; Falconi, M. Basophil recruitment into tumor-draining lymph nodes correlates with Th2 inflammation and reduced survival in pancreatic cancer patients. Cancer Res. 2016, 76, 1792–1803. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Cheng, P.; Condamine, T.; Mony, S.; Languino, L.R.; McCaffrey, J.C.; Hockstein, N.; Guarino, M.; Masters, G.; Penman, E. CD45 phosphatase inhibits STAT3 transcription factor activity in myeloid cells and promotes tumor-associated macrophage differentiation. Immunity 2016, 44, 303–315. [Google Scholar] [CrossRef] [Green Version]
- Franklin, R.A.; Liao, W.; Sarkar, A.; Kim, M.V.; Bivona, M.R.; Liu, K.; Pamer, E.G.; Li, M.O. The cellular and molecular origin of tumor-associated macrophages. Science 2014, 344, 921–925. [Google Scholar] [CrossRef] [Green Version]
- Sangaletti, S.; Di Carlo, E.; Gariboldi, S.; Miotti, S.; Cappetti, B.; Parenza, M.; Rumio, C.; Brekken, R.A.; Chiodoni, C.; Colombo, M.P. Macrophage-derived SPARC bridges tumor cell-extracellular matrix interactions toward metastasis. Cancer Res. 2008, 68, 9050–9059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nucera, S.; Biziato, D.; De Palma, M. The interplay between macrophages and angiogenesis in development, tissue injury and regeneration. Int. J. Dev. Biol. 2011, 55, 495–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, R.M.; Adam, M.; Hammond, J.; Orr, L.; Turbide, C. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J. Biol. Chem. 1987, 262, 9412–9420. [Google Scholar]
- Kim, H.K.; Song, K.; Park, Y.; Kang, Y.; Lee, Y.; Lee, K.; Ryu, K.; Bae, J.; Kim, S. Elevated levels of circulating platelet microparticles, VEGF, IL-6 and RANTES in patients with gastric cancer: Possible role of a metastasis predictor. Eur. J. Cancer 2003, 39, 184–191. [Google Scholar] [CrossRef]
- Kharaziha, P.; Ceder, S.; Li, Q.; Panaretakis, T. Tumor cell-derived exosomes: A message in a bottle. Biochim. Et Biophys. Acta (Bba)-Rev. Cancer 2012, 1826, 103–111. [Google Scholar] [CrossRef]
- Peinado, H.; Alečković, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; García-Santos, G.; Ghajar, C.M. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883. [Google Scholar] [CrossRef] [Green Version]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423. [Google Scholar] [CrossRef]
- Zhang, H.-G.; Grizzle, W.E. Exosomes: A novel pathway of local and distant intercellular communication that facilitates the growth and metastasis of neoplastic lesions. Am. J. Pathol. 2014, 184, 28–41. [Google Scholar] [CrossRef] [Green Version]
- Gatalica, Z.; Snyder, C.; Maney, T.; Ghazalpour, A.; Holterman, D.A.; Xiao, N.; Overberg, P.; Rose, I.; Basu, G.D.; Vranic, S. Programmed cell death 1 (PD-1) and its ligand (PD-L1) in common cancers and their correlation with molecular cancer type. Cancer Epidemiol. Prev. Biomark. 2014, 23, 2965–2970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, A.; Wang, H.; Liu, Y.; Zhao, M.; Zhang, H.; Lu, Z.; Fang, Y.; Chen, X.; Liu, G. The prognostic value of PD-L1 expression for non-small cell lung cancer patients: A meta-analysis. Eur. J. Surg. Oncol. (Ejso) 2015, 41, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Muenst, S.; Schaerli, A.; Gao, F.; Däster, S.; Trella, E.; Droeser, R.; Muraro, M.; Zajac, P.; Zanetti, R.; Gillanders, W. Expression of programmed death ligand 1 (PD-L1) is associated with poor prognosis in human breast cancer. Breast Cancer Res. Treat. 2014, 146, 15–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuang, D.-M.; Zhao, Q.; Peng, C.; Xu, J.; Zhang, J.-P.; Wu, C.; Zheng, L. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J. Exp. Med. 2009, 206, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef] [Green Version]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef]
- Han, Y.; Yang, Y.; Chen, Z.; Jiang, Z.; Gu, Y.; Liu, Y.; Xu, S.; Lin, C.; Pan, Z.; Zhou, W. Human hepatocellular carcinoma-infiltrating CD4+ CD69+ Foxp3− regulatory T cell suppresses T cell response via membrane-bound TGF-β1. J. Mol. Med. 2014, 92, 539–550. [Google Scholar] [CrossRef]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E. Nivolumab versus docetaxel in advanced nonsquamous non–small-cell lung cancer. New Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L. Pembrolizumab for the treatment of non–small-cell lung cancer. New Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C. Improved survival with ipilimumab in patients with metastatic melanoma. New Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. New Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Schadendorf, D.; Hodi, F.S.; Robert, C.; Weber, J.S.; Margolin, K.; Hamid, O.; Patt, D.; Chen, T.-T.; Berman, D.M.; Wolchok, J.D. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J. Clin. Oncol. 2015, 33, 1889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, C.; Thomas, L.; Bondarenko, I.; O’Day, S.; Weber, J.; Garbe, C.; Lebbe, C.; Baurain, J.-F.; Testori, A.; Grob, J.-J. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. New Engl. J. Med. 2011, 364, 2517–2526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Darvin, P.; Toor, S.M.; Nair, V.S.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilar, M.; Bhuket, T.; Torres, S.; Liu, B.; Wong, R.J. Prevalence of the metabolic syndrome in the United States, 2003-2012. Jama 2015, 313, 1973–1974. [Google Scholar] [CrossRef]
- Ranasinghe, P.; Mathangasinghe, Y.; Jayawardena, R.; Hills, A.; Misra, A. Prevalence and trends of metabolic syndrome among adults in the asia-pacific region: A systematic review. Bmc Public Health 2017, 17, 101. [Google Scholar] [CrossRef] [Green Version]
- Dunkley, A.; Charles, K.; Gray, L.; Camosso-Stefinovic, J.; Davies, M.; Khunti, K. Effectiveness of interventions for reducing diabetes and cardiovascular disease risk in people with metabolic syndrome: Systematic review and mixed treatment comparison meta-analysis. Diabetesobesity Metab. 2012, 14, 616–625. [Google Scholar] [CrossRef]
- Lorenzo, C.; Okoloise, M.; Williams, K.; Stern, M.P.; Haffner, S.M. The metabolic syndrome as predictor of type 2 diabetes: The San Antonio heart study. Diabetes Care 2003, 26, 3153–3159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grundy, S.M. Metabolic syndrome: Connecting and reconciling cardiovascular and diabetes worlds. J. Am. Coll. Cardiol. 2006, 47, 1093–1100. [Google Scholar] [CrossRef] [Green Version]
- Wilson, P.W.; D’Agostino, R.B.; Parise, H.; Sullivan, L.; Meigs, J.B. Metabolic syndrome as a precursor of cardiovascular disease and type 2 diabetes mellitus. Circulation 2005, 112, 3066–3072. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Pandeya, N.; Byrnes, G.; Renehan, A.G.; Stevens, G.A.; Ezzati, M.; Ferlay, J.; Miranda, J.J.; Romieu, I.; Dikshit, R. Global burden of cancer attributable to high body-mass index in 2012: A population-based study. Lancet Oncol. 2015, 16, 36–46. [Google Scholar] [CrossRef]
- Calle, E.E.; Kaaks, R. Overweight, obesity and cancer: Epidemiological evidence and proposed mechanisms. Nat. Rev. Cancer 2004, 4, 579–591. [Google Scholar] [CrossRef]
- Pearson-Stuttard, J.; Zhou, B.; Kontis, V.; Bentham, J.; Gunter, M.J.; Ezzati, M. Retracted: Worldwide Burden of Cancer Attributable to Diabetes and High Body-Mass Index: A Comparative Risk Assessment; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Calle, E.E.; Rodriguez, C.; Walker-Thurmond, K.; Thun, M.J. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of US adults. New Engl. J. Med. 2003, 348, 1625–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barone, B.B.; Yeh, H.-C.; Snyder, C.F.; Peairs, K.S.; Stein, K.B.; Derr, R.L.; Wolff, A.C.; Brancati, F.L. Long-term all-cause mortality in cancer patients with preexisting diabetes mellitus: A systematic review and meta-analysis. Jama 2008, 300, 2754–2764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipscombe, L.L.; Goodwin, P.J.; Zinman, B.; McLaughlin, J.R.; Hux, J.E. The impact of diabetes on survival following breast cancer. Breast Cancer Res. Treat. 2008, 109, 389–395. [Google Scholar] [CrossRef]
- Chen, Y.; Wu, F.; Saito, E.; Lin, Y.; Song, M.; Luu, H.N.; Gupta, P.C.; Sawada, N.; Tamakoshi, A.; Shu, X.-O. Association between type 2 diabetes and risk of cancer mortality: A pooled analysis of over 771,000 individuals in the Asia Cohort Consortium. Diabetologia 2017, 60, 1022–1032. [Google Scholar] [CrossRef]
- Vigneri, P.; Frasca, F.; Sciacca, L.; Pandini, G.; Vigneri, R. Diabetes and cancer. Endocr. -Relat. Cancer 2009, 16, 1103–1123. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Catrina, S.-B.; Okamoto, K.; Pereira, T.; Brismar, K.; Poellinger, L. Hyperglycemia regulates hypoxia-inducible factor-1α protein stability and function. Diabetes 2004, 53, 3226–3232. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Li, H.; Giovannucci, E.; Mucci, L.; Qiu, W.; Nguyen, P.L.; Gaziano, J.M.; Pollak, M.; Stampfer, M.J. Prediagnostic body-mass index, plasma C-peptide concentration, and prostate cancer-specific mortality in men with prostate cancer: A long-term survival analysis. Lancet Oncol. 2008, 9, 1039–1047. [Google Scholar] [CrossRef] [Green Version]
- Wolpin, B.M.; Meyerhardt, J.A.; Chan, A.T.; Ng, K.; Chan, J.A.; Wu, K.; Pollak, M.N.; Giovannucci, E.L.; Fuchs, C.S. Insulin, the insulin-like growth factor axis, and mortality in patients with nonmetastatic colorectal cancer. J. Clin. Oncol. 2009, 27, 176. [Google Scholar] [CrossRef] [PubMed]
- Perseghin, G.; Calori, G.; Lattuada, G.; Ragogna, F.; Dugnani, E.; Garancini, M.P.; Crosignani, P.; Villa, M.; Bosi, E.; Ruotolo, G. Insulin resistance/hyperinsulinemia and cancer mortality: The Cremona study at the 15th year of follow-up. Acta Diabetol. 2012, 49, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, T.; Kajio, H.; Sugiyama, T. Association between hyperinsulinemia and increased risk of cancer death in nonobese and obese people: A population-based observational study. Int. J. Cancer 2017, 141, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Coppack, S.W. Pro-inflammatory cytokines and adipose tissue. Proc. Nutr. Soc. 2001, 60, 349–356. [Google Scholar] [CrossRef] [Green Version]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef]
- Pollak, M. Insulin and insulin-like growth factor signalling in neoplasia. Nat. Rev. Cancer 2008, 8, 915–928. [Google Scholar] [CrossRef]
- Brahmkhatri, V.P.; Prasanna, C.; Atreya, H.S. Insulin-like growth factor system in cancer: Novel targeted therapies. Biomed Res. Int. 2015, 2015. [Google Scholar] [CrossRef] [Green Version]
- Le Roith, D. Insulin-like growth factors. New Engl. J. Med. 1997, 336, 633–640. [Google Scholar] [CrossRef]
- Rinderknecht, E.; Humbel, R.E. The amino acid sequence of human insulin-like growth factor I and its structural homology with proinsulin. J. Biol. Chem. 1978, 253, 2769–2776. [Google Scholar]
- LeRoith, D.; Werner, H.; Beitner-Johnson, D.; Roberts, C.T., Jr. Molecular and cellular aspects of the insulin-like growth factor I receptor. Endocr. Rev. 1995, 16, 143–163. [Google Scholar] [CrossRef]
- Kim, J.J.; Accili, D. Signalling through IGF-I and insulin receptors: Where is the specificity? Growth Horm. Igf Res. 2002, 12, 84–90. [Google Scholar] [CrossRef]
- Chitnis, M.M.; Yuen, J.S.; Protheroe, A.S.; Pollak, M.; Macaulay, V.M. The type 1 insulin-like growth factor receptor pathway. Clin. Cancer Res. 2008, 14, 6364–6370. [Google Scholar] [CrossRef] [Green Version]
- Hakuno, F.; Takahashi, S.-I. 40 years of IGF1: IGF1 receptor signaling pathways. J. Mol. Endocrinol. 2018, 61, T69–T86. [Google Scholar] [CrossRef] [Green Version]
- Robey, R.B.; Hay, N. Is Akt the “Warburg kinase”?—Akt-energy metabolism interactions and oncogenesis. Semin. Cancer Biol. 2009, 19, 25–31. [Google Scholar] [CrossRef] [Green Version]
- Cully, M.; You, H.; Levine, A.J.; Mak, T.W. Beyond PTEN mutations: The PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat. Rev. Cancer 2006, 6, 184–192. [Google Scholar] [CrossRef]
- Dai, N.; Ji, F.; Wright, J.; Minichiello, L.; Sadreyev, R.; Avruch, J. IGF2 mRNA binding protein-2 is a tumor promoter that drives cancer proliferation through its client mRNAs IGF2 and HMGA1. Elife 2017, 6, e27155. [Google Scholar] [CrossRef]
- Shukla, A.; Grisouard, J.; Ehemann, V.; Hermani, A.; Enzmann, H.; Mayer, D. Analysis of signaling pathways related to cell proliferation stimulated by insulin analogs in human mammary epithelial cell lines. Endocr. -Relat. Cancer 2009, 16, 429–441. [Google Scholar] [CrossRef] [Green Version]
- Shen, K.; Cui, D.; Sun, L.; Lu, Y.; Han, M.; Liu, J. Inhibition of IGF-IR increases chemosensitivity in human colorectal cancer cells through MRP-2 promoter suppression. J. Cell. Biochem. 2012, 113, 2086–2097. [Google Scholar] [CrossRef]
- Rochester, M.A.; Riedemann, J.; Hellawell, G.O.; Brewster, S.F.; Macaulay, V.M. Silencing of the IGF1R gene enhances sensitivity to DNA-damaging agents in both PTEN wild-type and mutant human prostate cancer. Cancer Gene Ther. 2005, 12, 90–100. [Google Scholar] [CrossRef]
- Chitnis, M.M.; Lodhia, K.A.; Aleksic, T.; Gao, S.; Protheroe, A.S.; Macaulay, V.M. IGF-1R inhibition enhances radiosensitivity and delays double-strand break repair by both non-homologous end-joining and homologous recombination. Oncogene 2014, 33, 5262–5273. [Google Scholar] [CrossRef] [Green Version]
- Ireland, L.; Santos, A.; Ahmed, M.S.; Rainer, C.; Nielsen, S.R.; Quaranta, V.; Weyer-Czernilofsky, U.; Engle, D.D.; Perez-Mancera, P.A.; Coupland, S.E. Chemoresistance in pancreatic cancer is driven by stroma-derived insulin-like growth factors. Cancer Res. 2016, 76, 6851–6863. [Google Scholar] [CrossRef] [Green Version]
- Nowak-Sliwinska, P.; van Beijnum, J.R.; Huijbers, E.J.; Gasull, P.C.; Mans, L.; Bex, A.; Griffioen, A.W. Oncofoetal insulin receptor isoform A marks the tumour endothelium; an underestimated pathway during tumour angiogenesis and angiostatic treatment. Br. J. Cancer 2019, 120, 218–228. [Google Scholar] [CrossRef] [Green Version]
- Ooi, G.T.; Tseng, L.; Tran, M.Q.; Rechler, M.M. Insulin rapidly decreases insulin-like growth factor-binding protein-1 gene transcription in streptozotocin-diabetic rats. Mol. Endocrinol. 1992, 6, 2219–2228. [Google Scholar]
- Powell, D.R.; Suwanichkul, A.; Cubbage, M.L.; DePaolis, L.A.; Snuggs, M.B.; Lee, P. Insulin inhibits transcription of the human gene for insulin-like growth factor-binding protein-1. J. Biol. Chem. 1991, 266, 18868–18876. [Google Scholar]
- Renehan, A.G.; Frystyk, J.; Flyvbjerg, A. Obesity and cancer risk: The role of the insulin–IGF axis. Trends Endocrinol. Metab. 2006, 17, 328–336. [Google Scholar] [CrossRef]
- Weinstein, D.; Simon, M.; Yehezkel, E.; Laron, Z.; Werner, H. Insulin analogues display IGF-I-like mitogenic and anti-apoptotic activities in cultured cancer cells. Diabetes/Metab. Res. Rev. 2009, 25, 41–49. [Google Scholar] [CrossRef]
- van Niekerk, G.; Christowitz, C.; Conradie, D.; Engelbrecht, A.-M. Insulin as an immunomodulatory hormone. Cytokine Growth Factor Rev. 2019. [Google Scholar] [CrossRef]
- Wensveen, F.M.; Šestan, M.; Turk Wensveen, T.; Polić, B. ‘Beauty and the beast’in infection: How immune–endocrine interactions regulate systemic metabolism in the context of infection. Eur. J. Immunol. 2019, 49, 982–995. [Google Scholar] [CrossRef] [Green Version]
- Bilbao, D.; Luciani, L.; Johannesson, B.; Piszczek, A.; Rosenthal, N. Insulin-like growth factor-1 stimulates regulatory T cells and suppresses autoimmune disease. Embo Mol. Med. 2014, 6, 1423–1435. [Google Scholar] [CrossRef]
- Johannesson, B.; Sattler, S.; Semenova, E.; Pastore, S.; Kennedy-Lydon, T.M.; Sampson, R.D.; Schneider, M.D.; Rosenthal, N.; Bilbao, D. Insulin-like growth factor-1 induces regulatory T cell-mediated suppression of allergic contact dermatitis in mice. Dis. Models Mech. 2014, 7, 977–985. [Google Scholar] [CrossRef] [Green Version]
- Miyagawa, I.; Nakayamada, S.; Nakano, K.; Yamagata, K.; Sakata, K.; Yamaoka, K.; Tanaka, Y. Induction of regulatory T cells and its regulation with insulin-like growth factor/insulin-like growth factor binding protein-4 by human mesenchymal stem cells. J. Immunol. 2017, 199, 1616–1625. [Google Scholar] [CrossRef] [Green Version]
- Kooijman, R.; Coppens, A. Insulin-like growth factor-I stimulates IL-10 production in human T cells. J. Leukoc. Biol. 2004, 76, 862–867. [Google Scholar] [CrossRef]
- Lu, M.C.; Yu, C.L.; Chen, H.C.; Yu, H.C.; Huang, H.B.; Lai, N.S. Increased miR-223 expression in T cells from patients with rheumatoid arthritis leads to decreased insulin-like growth factor-1-mediated interleukin-10 production. Clin. Exp. Immunol. 2014, 177, 641–651. [Google Scholar] [CrossRef]
- Barrett, J.P.; Minogue, A.M.; Falvey, A.; Lynch, M.A. Involvement of IGF-1 and Akt in M1/M2 activation state in bone marrow-derived macrophages. Exp. Cell Res. 2015, 335, 258–268. [Google Scholar] [CrossRef]
- Schabbauer, G.; Sharif, O.; Brunner, J.S.; Vogel, A. Macrophage rewiring by nutrient associated PI3K dependent pathways. Front. Immunol. 2019, 10, 2002. [Google Scholar]
- Ma, J.; Giovannucci, E.; Pollak, M.; Leavitt, A.; Tao, Y.; Gaziano, J.M.; Stampfer, M.J. A prospective study of plasma C-peptide and colorectal cancer risk in men. J. Natl. Cancer Inst. 2004, 96, 546–553. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, P.J.; Ennis, M.; Pritchard, K.I.; Trudeau, M.E.; Koo, J.; Madarnas, Y.; Hartwick, W.; Hoffman, B.; Hood, N. Fasting insulin and outcome in early-stage breast cancer: Results of a prospective cohort study. J. Clin. Oncol. 2002, 20, 42–51. [Google Scholar] [CrossRef]
- Cummings, D.E.; Overduin, J.; Foster-Schubert, K.E. Gastric bypass for obesity: Mechanisms of weight loss and diabetes resolution. J. Clin. Endocrinol. Metab. 2004, 89, 2608–2615. [Google Scholar] [CrossRef]
- Lau, D.C.; Teoh, H. Benefits of modest weight loss on the management of type 2 diabetes mellitus. Can. J. Diabetes 2013, 37, 128–134. [Google Scholar] [CrossRef]
- Pendyala, S.; Neff, L.M.; Suarez-Farinas, M.; Holt, P.R. Diet-induced weight loss reduces colorectal inflammation: Implications for colorectal carcinogenesis. Am. J. Clin. Nutr. 2011, 93, 234–242. [Google Scholar] [CrossRef] [Green Version]
- Rapp, K.; Klenk, J.; Ulmer, H.; Concin, H.; Diem, G.; Oberaigner, W.; Schroeder, J. Weight change and cancer risk in a cohort of more than 65 000 adults in Austria. Ann. Oncol. 2008, 19, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Sjöström, L.; Gummesson, A.; Sjöström, C.D.; Narbro, K.; Peltonen, M.; Wedel, H.; Bengtsson, C.; Bouchard, C.; Carlsson, B.; Dahlgren, S. Effects of bariatric surgery on cancer incidence in obese patients in Sweden (Swedish Obese Subjects Study): A prospective, controlled intervention trial. Lancet Oncol. 2009, 10, 653–662. [Google Scholar] [CrossRef]
- Bai, Y.; Sun, Q. Macrophage recruitment in obese adipose tissue. Obes. Rev. 2015, 16, 127–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, E.J.; Lee, J.H.; Yu, G.-Y.; He, G.; Ali, S.R.; Holzer, R.G.; Österreicher, C.H.; Takahashi, H.; Karin, M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 2010, 140, 197–208. [Google Scholar] [CrossRef] [Green Version]
- Ceccarini, G.; Pelosini, C.; Ferrari, F.; Magno, S.; Vitti, J.; Salvetti, G.; Moretto, C.; Marioni, A.; Buccianti, P.; Piaggi, P. Serum IGF-binding protein 2 (IGFBP-2) concentrations change early after gastric bypass bariatric surgery revealing a possible marker of leptin sensitivity in obese subjects. Endocrine 2019, 65, 86–93. [Google Scholar] [CrossRef]
- Bailey, C.J. Metformin: Historical overview. Diabetologia 2017, 60, 1566–1576. [Google Scholar] [CrossRef] [Green Version]
- Kay, J.P.; Alemzadeh, R.; Langley, G.; D’angelo, L.; Smith, P.; Holshouser, S. Beneficial effects of metformin in normoglycemic morbidly obese adolescents. Metab. -Clin. Exp. 2001, 50, 1457–1461. [Google Scholar] [CrossRef]
- Fontbonne, A.; Charles, M.A.; Juhan-Vague, I.; Bard, J.M.; André, P.; Isnard, F.; Cohen, J.M.; Grandmottet, P.; Vague, P.; Safar, M.E. The effect of metformin on the metabolic abnormalities associated with upper-body fat distribution. BIGPRO Study Group. Diabetes Care 1996, 19, 920–926. [Google Scholar] [CrossRef]
- Zakikhani, M.; Dowling, R.; Fantus, I.G.; Sonenberg, N.; Pollak, M. Metformin is an AMP kinase–dependent growth inhibitor for breast cancer cells. Cancer Res. 2006, 66, 10269–10273. [Google Scholar] [CrossRef] [Green Version]
- Alimova, I.N.; Liu, B.; Fan, Z.; Edgerton, S.M.; Dillon, T.; Lind, S.E.; Thor, A.D. Metformin inhibits breast cancer cell growth, colony formation and induces cell cycle arrest in vitro. Cell Cycle 2009, 8, 909–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Fan, Z.; Edgerton, S.M.; Deng, X.-S.; Alimova, I.N.; Lind, S.E.; Thor, A.D. Metformin induces unique biological and molecular responses in triple negative breast cancer cells. Cell Cycle 2009, 8, 2031–2040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowling, R.J.; Zakikhani, M.; Fantus, I.G.; Pollak, M.; Sonenberg, N. Metformin inhibits mammalian target of rapamycin–dependent translation initiation in breast cancer cells. Cancer Res. 2007, 67, 10804–10812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Algire, C.; Zakikhani, M.; Blouin, M.-J.; Shuai, J.H.; Pollak, M. Metformin attenuates the stimulatory effect of a high-energy diet on in vivo LLC1 carcinoma growth. Endocr. -Relat. Cancer 2008, 15, 833–839. [Google Scholar] [CrossRef]
- Algire, C.; Amrein, L.; Bazile, M.; David, S.; Zakikhani, M.; Pollak, M. Diet and tumor LKB1 expression interact to determine sensitivity to anti-neoplastic effects of metformin in vivo. Oncogene 2011, 30, 1174–1182. [Google Scholar] [CrossRef] [Green Version]
- Algire, C.; Amrein, L.; Zakikhani, M.; Panasci, L.; Pollak, M. Metformin blocks the stimulative effect of a high-energy diet on colon carcinoma growth in vivo and is associated with reduced expression of fatty acid synthase. Endocr. -Relat. Cancer 2010, 17, 351. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, E.J.; LeRoith, D. Minireview: IGF, insulin, and cancer. Endocrinology 2011, 152, 2546–2551. [Google Scholar] [CrossRef] [Green Version]
- Tsugane, S.; Inoue, M. Insulin resistance and cancer: Epidemiological evidence. Cancer Sci. 2010, 101, 1073–1079. [Google Scholar] [CrossRef]
- Evans, J.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. Bmj 2005, 330, 1304–1305. [Google Scholar] [CrossRef] [Green Version]
- Bowker, S.L.; Majumdar, S.R.; Veugelers, P.; Johnson, J.A. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diabetes Care 2006, 29, 254–258. [Google Scholar] [CrossRef] [Green Version]
- Currie, C.; Poole, C.; Gale, E. The influence of glucose-lowering therapies on cancer risk in type 2 diabetes. Diabetologia 2009, 52, 1766–1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monami, M.; Lamanna, C.; Balzi, D.; Marchionni, N.; Mannucci, E. Sulphonylureas and cancer: A case–control study. Acta Diabetol. 2009, 46, 279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, J.L.; Stanford, J.L. Metformin use and prostate cancer in Caucasian men: Results from a population-based case–control study. Cancer Causes Control 2009, 20, 1617. [Google Scholar] [CrossRef] [Green Version]
- Landman, G.W.; Kleefstra, N.; van Hateren, K.J.; Groenier, K.H.; Gans, R.O.; Bilo, H.J. Metformin associated with lower cancer mortality in type 2 diabetes: ZODIAC-16. Diabetes Care 2010, 33, 322–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonasson, J.; Ljung, R.; Talbäck, M.; Haglund, B.; Gudbjörnsdòttir, S.; Steineck, G. Insulin glargine use and short-term incidence of malignancies—a population-based follow-up study in Sweden. Diabetologia 2009, 52, 1745–1754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colhoun, H. Use of insulin glargine and cancer incidence in Scotland: A study from the Scottish Diabetes Research Network Epidemiology Group. Diabetologia 2009, 52, 1755–1765. [Google Scholar] [CrossRef] [Green Version]
- Hemkens, L.G.; Grouven, U.; Bender, R.; Günster, C.; Gutschmidt, S.; Selke, G.W.; Sawicki, P.T. Risk of malignancies in patients with diabetes treated with human insulin or insulin analogues: A cohort study. Diabetologia 2009, 52, 1732–1744. [Google Scholar] [CrossRef] [Green Version]
- DeCensi, A.; Puntoni, M.; Goodwin, P.; Cazzaniga, M.; Gennari, A.; Bonanni, B.; Gandini, S. Metformin and cancer risk in diabetic patients: A systematic review and meta-analysis. Cancer Prev. Res. 2010, 3, 1451–1461. [Google Scholar] [CrossRef] [Green Version]
- Jabbour, M. Primary care physicians and insulin initiation: Multiple barriers, lack of knowledge or both? Int. J. Clin. Pract. 2008, 62, 845–847. [Google Scholar] [CrossRef] [Green Version]
- Kurtzhals, P.; Schäffer, L.; Sørensen, A.; Kristensen, C.; Jonassen, I.; Schmid, C.; Trüb, T. Correlations of receptor binding and metabolic and mitogenic potencies of insulin analogs designed for clinical use. Diabetes 2000, 49, 999–1005. [Google Scholar] [CrossRef] [Green Version]
- Liefvendahl, E.; Arnqvist, H. Mitogenic effect of the insulin analogue glargine in malignant cells in comparison with insulin and IGF-I. Horm. Metab. Res. 2008, 40, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Cheong, J.-H.; Park, E.S.; Liang, J.; Dennison, J.B.; Tsavachidou, D.; Nguyen-Charles, C.; Cheng, K.W.; Hall, H.; Zhang, D.; Lu, Y. Dual inhibition of tumor energy pathway by 2-deoxyglucose and metformin is effective against a broad spectrum of preclinical cancer models. Mol. Cancer Ther. 2011, 10, 2350–2362. [Google Scholar] [CrossRef] [Green Version]
- Sahra, I.B.; Laurent, K.; Giuliano, S.; Larbret, F.; Ponzio, G.; Gounon, P.; Le Marchand-Brustel, Y.; Giorgetti-Peraldi, S.; Cormont, M.; Bertolotto, C. Targeting cancer cell metabolism: The combination of metformin and 2-deoxyglucose induces p53-dependent apoptosis in prostate cancer cells. Cancer Res. 2010, 70, 2465–2475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buzzai, M.; Jones, R.G.; Amaravadi, R.K.; Lum, J.J.; DeBerardinis, R.J.; Zhao, F.; Viollet, B.; Thompson, C.B. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007, 67, 6745–6752. [Google Scholar] [CrossRef] [Green Version]
- Yap, T.A.; Bjerke, L.; Clarke, P.A.; Workman, P. Drugging PI3K in cancer: Refining targets and therapeutic strategies. Curr. Opin. Pharmacol. 2015, 23, 98–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, C.X.; Sanchez, C.; Gao, F.; Crowder, R.; Naughton, M.; Pluard, T.; Creekmore, A.; Guo, Z.; Hoog, J.; Lockhart, A.C. A phase I study of the AKT inhibitor MK-2206 in combination with hormonal therapy in postmenopausal women with estrogen receptor–positive metastatic breast cancer. Clin. Cancer Res. 2016, 22, 2650–2658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sprinzl, M.F.; Puschnik, A.; Schlitter, A.M.; Schad, A.; Ackermann, K.; Esposito, I.; Lang, H.; Galle, P.R.; Weinmann, A.; Heikenwälder, M. Sorafenib inhibits macrophage-induced growth of hepatoma cells by interference with insulin-like growth factor-1 secretion. J. Hepatol. 2015, 62, 863–870. [Google Scholar] [CrossRef]
- Bowers, L.W.; Rossi, E.L.; O’Flanagan, C.H.; deGraffenried, L.A.; Hursting, S.D. The role of the insulin/IGF system in cancer: Lessons learned from clinical trials and the energy balance-cancer link. Front. Endocrinol. 2015, 6, 77. [Google Scholar] [CrossRef] [Green Version]
- Simpson, A.; Petnga, W.; Macaulay, V.M.; Weyer-Czernilofsky, U.; Bogenrieder, T. Insulin-like growth factor (IGF) pathway targeting in cancer: Role of the IGF axis and opportunities for future combination studies. Target. Oncol. 2017, 12, 571–597. [Google Scholar] [CrossRef] [Green Version]
- Pollak, M. The insulin and insulin-like growth factor receptor family in neoplasia: An update. Nat. Rev. Cancer 2012, 12, 159–169. [Google Scholar] [CrossRef]
- Busaidy, N.L.; Farooki, A.; Dowlati, A.; Perentesis, J.P.; Dancey, J.E.; Doyle, L.A.; Brell, J.M.; Siu, L.L. Management of metabolic effects associated with anticancer agents targeting the PI3K-Akt-mTOR pathway. J. Clin. Oncol. 2012, 30, 2919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, H.J.; Sie, C.; Schumann, E.; Witte, A.-K.; Dressel, R.; van den Brandt, J.; Reichardt, H.M. The insulin receptor plays a critical role in T cell function and adaptive immunity. J. Immunol. 2017, 198, 1910–1920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quail, D.F.; Bowman, R.L.; Akkari, L.; Quick, M.L.; Schuhmacher, A.J.; Huse, J.T.; Holland, E.C.; Sutton, J.C.; Joyce, J.A. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science 2016, 352, aad3018. [Google Scholar] [CrossRef] [Green Version]
- Andrews, D.W.; Resnicoff, M.; Flanders, A.E.; Kenyon, L.; Curtis, M.; Merli, G.; Baserga, R.; Iliakis, G.; Aiken, R.D. Results of a pilot study involving the use of an antisense oligodeoxynucleotide directed against the insulin-like growth factor type I receptor in malignant astrocytomas. J. Clin. Oncol. 2001, 19, 2189–2200. [Google Scholar] [CrossRef] [PubMed]
- Resnicoff, M.; Sell, C.; Rubini, M.; Coppola, D.; Ambrose, D.; Baserga, R.; Rubin, R. Rat glioblastoma cells expressing an antisense RNA to the insulin-like growth factor-1 (IGF-1) receptor are nontumorigenic and induce regression of wild-type tumors. Cancer Res. 1994, 54, 2218–2222. [Google Scholar] [PubMed]
- Trojan, J.; Johnson, T.R.; Rudin, S.D.; Ilan, J.; Tykocinski, M.L. Treatment and prevention of rat glioblastoma by immunogenic C6 cells expressing antisense insulin-like growth factor I RNA. Science 1993, 259, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Somasundaram, R.; Zhang, G.; Wagner, S.N.; Fukunaga-Kalabis, M.; Herlyn, M. The role of tumor microenvironment in therapy resistance and melanoma progression. AACR 2015. [Google Scholar]
- Afzal, M.Z.; Mercado, R.R.; Shirai, K. Efficacy of metformin in combination with immune checkpoint inhibitors (anti-PD-1/anti-CTLA-4) in metastatic malignant melanoma. J. Immunother. Cancer 2018, 6, 64. [Google Scholar] [CrossRef] [Green Version]
- Pietras, R.; Xu, H.; Hu, X.; Matheny, C.; Sandler, A.; Patel, M. P1. 04-33 retrospective descriptive analysis of metformin with atezolizumab in advanced non-small cell lung cancer in the OAK trial. J. Thorac. Oncol. 2018, 13, S538–S539. [Google Scholar] [CrossRef] [Green Version]
- Bahrambeigi, S.; Shafiei-Irannejad, V. Immune-mediated anti-tumor effects of metformin; targeting metabolic reprogramming of T cells as a new possible mechanism for anti-cancer effects of metformin. Biochem. Pharmacol. 2020, 174, 113787. [Google Scholar] [CrossRef]
- Araki, K.; Turner, A.P.; Shaffer, V.O.; Gangappa, S.; Keller, S.A.; Bachmann, M.F.; Larsen, C.P.; Ahmed, R. mTOR regulates memory CD8 T-cell differentiation. Nature 2009, 460, 108–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prlic, M.; Bevan, M.J. A metabolic switch to memory. Nature 2009, 460, 41–42. [Google Scholar] [CrossRef] [PubMed]
- Curry, J.M.; Johnson, J.; Mollaee, M.; Tassone, P.; Amin, D.; Knops, A.; Whitaker-Menezes, D.; Mahoney, M.G.; South, A.; Rodeck, U. Metformin Clinical Trial in HPV+ and HPV–Head and Neck Squamous Cell Carcinoma: Impact on Cancer Cell Apoptosis and Immune Infiltrate. Front. Oncol. 2018, 8, 436. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Liang, G.; Yao, Z.; Zhang, J.; Liu, R.; Chen, H.; Zhou, Y.; Wu, H.; Yang, B.; He, Q. Metformin prevents cancer metastasis by inhibiting M2-like polarization of tumor associated macrophages. Oncotarget 2015, 6, 36441. [Google Scholar] [CrossRef] [Green Version]
- Uehara, T.; Eikawa, S.; Nishida, M.; Kunisada, Y.; Yoshida, A.; Fujiwara, T.; Kunisada, T.; Ozaki, T.; Udono, H. Metformin induces CD11b+-cell-mediated growth inhibition of an osteosarcoma: Implications for metabolic reprogramming of myeloid cells and anti-tumor effects. Int. Immunol. 2019, 31, 187–198. [Google Scholar] [CrossRef] [Green Version]
- Verdura, S.; Cuyàs, E.; Martin-Castillo, B.; Menendez, J.A. Metformin as an archetype immuno-metabolic adjuvant for cancer immunotherapy. OncoImmunology 2019, 8, e1633235. [Google Scholar] [CrossRef]
- Cha, J.-H.; Yang, W.-H.; Xia, W.; Wei, Y.; Chan, L.-C.; Lim, S.-O.; Li, C.-W.; Kim, T.; Chang, S.-S.; Lee, H.-H. Metformin promotes antitumor immunity via endoplasmic-reticulum-associated degradation of PD-L1. Mol. Cell 2018, 71, 606–620.e607. [Google Scholar] [CrossRef] [Green Version]
- Garner, H.; de Visser, K.E. Immune crosstalk in cancer progression and metastatic spread: A complex conversation. Nat. Rev. Immunol. 2020, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palucka, A.K.; Coussens, L.M. The basis of oncoimmunology. Cell 2016, 164, 1233–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elgert, K.D.; Alleva, D.G.; Mullins, D.W. Tumor-induced immune dysfunction: The macrophage connection. J. Leukoc. Biol. 1998, 64, 275–290. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Jiang, A. Dendritic cells and CD8 T cell immunity in tumor microenvironment. Front. Immunol. 2018, 9, 3059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, C.E.; Pollard, J.W. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006, 66, 605–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almand, B.; Resser, J.R.; Lindman, B.; Nadaf, S.; Clark, J.I.; Kwon, E.D.; Carbone, D.P.; Gabrilovich, D.I. Clinical significance of defective dendritic cell differentiation in cancer. Clin. Cancer Res. 2000, 6, 1755–1766. [Google Scholar] [PubMed]
- Tang, M.; Diao, J.; Cattral, M.S. Molecular mechanisms involved in dendritic cell dysfunction in cancer. Cell. Mol. Life Sci. 2017, 74, 761–776. [Google Scholar] [CrossRef]
- Xu, M.M.; Pu, Y.; Han, D.; Shi, Y.; Cao, X.; Liang, H.; Chen, X.; Li, X.-D.; Deng, L.; Chen, Z.J. Dendritic cells but not macrophages sense tumor mitochondrial DNA for cross-priming through signal regulatory protein α signaling. Immunity 2017, 47, 363–373.e365. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting edge: Distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 2011, 186, 3299–3303. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.Z.; Wang, R.; Huang, G.; Vogel, P.; Neale, G.; Green, D.R.; Chi, H. HIF1α–dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J. Exp. Med. 2011, 208, 1367–1376. [Google Scholar] [CrossRef] [Green Version]
- Gubser, P.M.; Bantug, G.R.; Razik, L.; Fischer, M.; Dimeloe, S.; Hoenger, G.; Durovic, B.; Jauch, A.; Hess, C. Rapid effector function of memory CD8+ T cells requires an immediate-early glycolytic switch. Nat. Immunol. 2013, 14, 1064. [Google Scholar] [CrossRef]
- Dang, E.V.; Barbi, J.; Yang, H.-Y.; Jinasena, D.; Yu, H.; Zheng, Y.; Bordman, Z.; Fu, J.; Kim, Y.; Yen, H.-R. Control of TH17/Treg balance by hypoxia-inducible factor 1. Cell 2011, 146, 772–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Prados, J.-C.; Través, P.G.; Cuenca, J.; Rico, D.; Aragonés, J.; Martín-Sanz, P.; Cascante, M.; Boscá, L. Substrate fate in activated macrophages: A comparison between innate, classic, and alternative activation. J. Immunol. 2010, 185, 605–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krawczyk, C.M.; Holowka, T.; Sun, J.; Blagih, J.; Amiel, E.; DeBerardinis, R.J.; Cross, J.R.; Jung, E.; Thompson, C.B.; Jones, R.G. Toll-like receptor–induced changes in glycolytic metabolism regulate dendritic cell activation. Bloodthe J. Am. Soc. Hematol. 2010, 115, 4742–4749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.-H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; Van Der Windt, G.J. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, P.-C.; Bihuniak, J.D.; Macintyre, A.N.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.-C.; Cui, G.; Micevic, G.; Perales, J.C. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell 2015, 162, 1217–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottfried, E.; Kunz-Schughart, L.A.; Ebner, S.; Mueller-Klieser, W.; Hoves, S.; Andreesen, R.; Mackensen, A.; Kreutz, M. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood 2006, 107, 2013–2021. [Google Scholar] [CrossRef]
- Dietl, K.; Renner, K.; Dettmer, K.; Timischl, B.; Eberhart, K.; Dorn, C.; Hellerbrand, C.; Kastenberger, M.; Kunz-Schughart, L.A.; Oefner, P.J. Lactic acid and acidification inhibit TNF secretion and glycolysis of human monocytes. J. Immunol. 2010, 184, 1200–1209. [Google Scholar] [CrossRef]
- Colegio, O.R.; Chu, N.-Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- Mukhopadhyay, R.; Jia, J.; Arif, A.; Ray, P.S.; Fox, P.L. The GAIT system: A gatekeeper of inflammatory gene expression. Trends Biochem. Sci. 2009, 34, 324–331. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-H.; Curtis, J.D.; Maggi Jr, L.B.; Faubert, B.; Villarino, A.V.; O’Sullivan, D.; Huang, S.C.-C.; Van Der Windt, G.J.; Blagih, J.; Qiu, J. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 2013, 153, 1239–1251. [Google Scholar] [CrossRef] [Green Version]
- Sukumar, M.; Liu, J.; Ji, Y.; Subramanian, M.; Crompton, J.G.; Yu, Z.; Roychoudhuri, R.; Palmer, D.C.; Muranski, P.; Karoly, E.D. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J. Clin. Investig. 2013, 123, 4479–4488. [Google Scholar] [CrossRef] [PubMed]
- Pollizzi, K.N.; Patel, C.H.; Sun, I.-H.; Oh, M.-H.; Waickman, A.T.; Wen, J.; Delgoffe, G.M.; Powell, J.D. mTORC1 and mTORC2 selectively regulate CD8+ T cell differentiation. J. Clin. Investig. 2015, 125, 2090–2108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, A.K.; Huang, S.C.-C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerriets, V.A.; Kishton, R.J.; Nichols, A.G.; Macintyre, A.N.; Inoue, M.; Ilkayeva, O.; Winter, P.S.; Liu, X.; Priyadharshini, B.; Slawinska, M.E. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J. Clin. Investig. 2015, 125, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Beier, U.H.; Angelin, A.; Akimova, T.; Wang, L.; Liu, Y.; Xiao, H.; Koike, M.A.; Hancock, S.A.; Bhatti, T.R.; Han, R. Essential role of mitochondrial energy metabolism in Foxp3+ T-regulatory cell function and allograft survival. Faseb J. 2015, 29, 2315–2326. [Google Scholar] [CrossRef] [Green Version]
- Procaccini, C.; Carbone, F.; Di Silvestre, D.; Brambilla, F.; De Rosa, V.; Galgani, M.; Faicchia, D.; Marone, G.; Tramontano, D.; Corona, M. The proteomic landscape of human ex vivo regulatory and conventional T cells reveals specific metabolic requirements. Immunity 2016, 44, 406–421. [Google Scholar] [CrossRef] [Green Version]
- O’Sullivan, D.; van der Windt, G.J.; Huang, S.C.-C.; Curtis, J.D.; Chang, C.-H.; Buck, M.D.; Qiu, J.; Smith, A.M.; Lam, W.Y.; DiPlato, L.M. Memory CD8+ T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity 2014, 41, 75–88. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, L.A.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553. [Google Scholar] [CrossRef] [Green Version]
- Molodtsov, A.; Turk, M.J. Tissue resident CD8 memory T cell responses in cancer and autoimmunity. Front. Immunol. 2018, 9, 2810. [Google Scholar] [CrossRef]
- Menares, E.; Gálvez-Cancino, F.; Cáceres-Morgado, P.; Ghorani, E.; López, E.; Díaz, X.; Saavedra-Almarza, J.; Figueroa, D.A.; Roa, E.; Quezada, S.A. Tissue-resident memory CD8+ T cells amplify anti-tumor immunity by triggering antigen spreading through dendritic cells. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- van der Windt, G.J.; O’Sullivan, D.; Everts, B.; Huang, S.C.-C.; Buck, M.D.; Curtis, J.D.; Chang, C.-H.; Smith, A.M.; Ai, T.; Faubert, B. CD8 memory T cells have a bioenergetic advantage that underlies their rapid recall ability. Proc. Natl. Acad. Sci. USA 2013, 110, 14336–14341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Windt, G.J.; Everts, B.; Chang, C.-H.; Curtis, J.D.; Freitas, T.C.; Amiel, E.; Pearce, E.J.; Pearce, E.L. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity 2012, 36, 68–78. [Google Scholar] [CrossRef] [Green Version]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese Jr, R.V. Cellular fatty acid metabolism and cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Svensson, R.U.; Parker, S.J.; Eichner, L.J.; Kolar, M.J.; Wallace, M.; Brun, S.N.; Lombardo, P.S.; Van Nostrand, J.L.; Hutchins, A.; Vera, L. Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nat. Med. 2016, 22, 1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flaveny, C.A.; Griffett, K.; El-Gendy, B.E.-D.M.; Kazantzis, M.; Sengupta, M.; Amelio, A.L.; Chatterjee, A.; Walker, J.; Solt, L.A.; Kamenecka, T.M. Broad anti-tumor activity of a small molecule that selectively targets the Warburg effect and lipogenesis. Cancer Cell 2015, 28, 42–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Everts, B.; Amiel, E.; Huang, S.C.-C.; Smith, A.M.; Chang, C.-H.; Lam, W.Y.; Redmann, V.; Freitas, T.C.; Blagih, J.; Van Der Windt, G.J. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKɛ supports the anabolic demands of dendritic cell activation. Nat. Immunol. 2014, 15, 323. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.W.; Heiniger, H.-J.; Kandutsch, A.A. Relationship between sterol synthesis and DNA synthesis in phytohemagglutinin-stimulated mouse lymphocytes. Proc. Natl. Acad. Sci. USA 1975, 72, 1950–1954. [Google Scholar] [CrossRef] [Green Version]
- Dufort, F.J.; Gumina, M.R.; Ta, N.L.; Tao, Y.; Heyse, S.A.; Scott, D.A.; Richardson, A.D.; Seyfried, T.N.; Chiles, T.C. Glucose-dependent de novo lipogenesis in B lymphocytes a requirement for ATP-citrate lyase in lipopolysaccharide-induced differentiation. J. Biol. Chem. 2014, 289, 7011–7024. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Walsh, M.C.; Hoehn, K.L.; James, D.E.; Wherry, E.J.; Choi, Y. Regulator of fatty acid metabolism, acetyl coenzyme a carboxylase 1, controls T cell immunity. J. Immunol. 2014, 192, 3190–3199. [Google Scholar] [CrossRef] [Green Version]
- Berod, L.; Friedrich, C.; Nandan, A.; Freitag, J.; Hagemann, S.; Harmrolfs, K.; Sandouk, A.; Hesse, C.; Castro, C.N.; Bähre, H. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat. Med. 2014, 20, 1327. [Google Scholar] [CrossRef]
- Gullino, P.M.; Clark, S.H.; Grantham, F.H. The interstitial fluid of solid tumors. Cancer Res. 1964, 24, 780–797. [Google Scholar] [PubMed]
- Buck, M.D.; O’Sullivan, D.; Geltink, R.I.K.; Curtis, J.D.; Chang, C.-H.; Sanin, D.E.; Qiu, J.; Kretz, O.; Braas, D.; van der Windt, G.J. Mitochondrial dynamics controls T cell fate through metabolic programming. Cell 2016, 166, 63–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menk, A.V.; Scharping, N.E.; Rivadeneira, D.B.; Calderon, M.J.; Watson, M.J.; Dunstane, D.; Watkins, S.C.; Delgoffe, G.M. 4-1BB costimulation induces T cell mitochondrial function and biogenesis enabling cancer immunotherapeutic responses. J. Exp. Med. 2018, 215, 1091–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teijeira, A.; Labiano, S.; Garasa, S.; Etxeberria, I.; Santamaría, E.; Rouzaut, A.; Enamorado, M.; Azpilikueta, A.; Inoges, S.; Bolaños, E. Mitochondrial morphological and functional reprogramming following CD137 (4-1BB) costimulation. Cancer Immunol. Res. 2018. [Google Scholar] [CrossRef] [Green Version]
- Cascone, T.; McKenzie, J.A.; Mbofung, R.M.; Punt, S.; Wang, Z.; Xu, C.; Williams, L.J.; Wang, Z.; Bristow, C.A.; Carugo, A. Increased tumor glycolysis characterizes immune resistance to adoptive T cell therapy. Cell Metab. 2018, 27, 977–987.e974. [Google Scholar] [CrossRef]
- Scharping, N.E.; Menk, A.V.; Whetstone, R.D.; Zeng, X.; Delgoffe, G.M. Efficacy of PD-1 blockade is potentiated by metformin-induced reduction of tumor hypoxia. Cancer Immunol. Res. 2017, 5, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Bai, Y.; Xiong, Y.; Zhang, J.; Chen, S.; Zheng, X.; Meng, X.; Li, L.; Wang, J.; Xu, C. Potentiating the antitumour response of CD8+ T cells by modulating cholesterol metabolism. Nature 2016, 531, 651–655. [Google Scholar] [CrossRef] [Green Version]
- Dean, E.J.; Falchook, G.S.; Patel, M.R.; Brenner, A.J.; Infante, J.R.; Arkenau, H.-T.; Borazanci, E.H.; Lopez, J.S.; Pant, S.; Schmid, P. Preliminary activity in the first in human study of the first-in-class fatty acid synthase (FASN) inhibitor, TVB-2640. Am. Soc. Clin. Oncol. 2016. [Google Scholar] [CrossRef]
- Hosios, A.M.; Hecht, V.C.; Danai, L.V.; Johnson, M.O.; Rathmell, J.C.; Steinhauser, M.L.; Manalis, S.R.; Vander Heiden, M.G. Amino acids rather than glucose account for the majority of cell mass in proliferating mammalian cells. Dev. Cell 2016, 36, 540–549. [Google Scholar] [CrossRef] [Green Version]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619. [Google Scholar] [CrossRef]
- Wang, R.; Dillon, C.P.; Shi, L.Z.; Milasta, S.; Carter, R.; Finkelstein, D.; McCormick, L.L.; Fitzgerald, P.; Chi, H.; Munger, J. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 2011, 35, 871–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carr, E.L.; Kelman, A.; Wu, G.S.; Gopaul, R.; Senkevitch, E.; Aghvanyan, A.; Turay, A.M.; Frauwirth, K.A. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J. Immunol. 2010, 185, 1037–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawford, J.; Cohen, H.J. The essential role of L-glutamine in lymphocyte differentiation in vitro. J. Cell. Physiol. 1985, 124, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Nakaya, M.; Xiao, Y.; Zhou, X.; Chang, J.-H.; Chang, M.; Cheng, X.; Blonska, M.; Lin, X.; Sun, S.-C. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity 2014, 40, 692–705. [Google Scholar] [CrossRef] [Green Version]
- Klysz, D.; Tai, X.; Robert, P.A.; Craveiro, M.; Cretenet, G.; Oburoglu, L.; Mongellaz, C.; Floess, S.; Fritz, V.; Matias, M.I. Glutamine-dependent α-ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci. Signal. 2015, 8, ra97. [Google Scholar] [CrossRef] [Green Version]
- Lemberg, K.M.; Vornov, J.J.; Rais, R.; Slusher, B.S. We’re not “DON” yet: Optimal dosing and prodrug delivery of 6-Diazo-5-oxo-L-norleucine. Mol. Cancer Ther. 2018, 17, 1824–1832. [Google Scholar] [CrossRef] [Green Version]
- Pinkus, L.M. [45] Glutamine binding sites. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1977; Volume 46, pp. 414–427. [Google Scholar]
- Magill, G.; Myers, W.; Reilly, H.; Putnam, R.; Magill, J.; Sykes, M.; Escher, G.; Karnofsky, D.; Burchenal, J. Pharmacological and initial therapeutic observations on 6-Diazo-5-Oxo-L-Norleucine (Don) in human neoplastic disease. Cancer 1957, 10, 1138–1150. [Google Scholar] [CrossRef]
- Leone, R.D.; Zhao, L.; Englert, J.M.; Sun, I.-M.; Oh, M.-H.; Sun, I.-H.; Arwood, M.L.; Bettencourt, I.A.; Patel, C.H.; Wen, J. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 2019, 366, 1013–1021. [Google Scholar] [CrossRef]
- Palmieri, E.M.; Menga, A.; Martín-Pérez, R.; Quinto, A.; Riera-Domingo, C.; De Tullio, G.; Hooper, D.C.; Lamers, W.H.; Ghesquière, B.; McVicar, D.W. Pharmacologic or genetic targeting of glutamine synthetase skews macrophages toward an M1-like phenotype and inhibits tumor metastasis. Cell Rep. 2017, 20, 1654–1666. [Google Scholar] [CrossRef] [Green Version]
- Oh, M.-H.; Sun, I.-H.; Zhao, L.; Leone, R.D.; Sun, I.-M.; Xu, W.; Collins, S.L.; Tam, A.J.; Blosser, R.L.; Patel, C.H. Targeting glutamine metabolism enhances tumor specific immunity by modulating suppressive myeloid cells. J. Clin. Investig. 2020. [Google Scholar] [CrossRef]
- Guerra, V.; Dinardo, C.D.; Konopleva, M.; Burger, J.A.; Borthakur, G.; Jabbour, E.; Pemmaraju, N.; Sheppard, K.; Garcia-Manero, G.; Kantarjian, H.M. Interim results from a phase Ib/II clinical study of the glutaminase inhibitor telaglenastat (CB-839) in combination with azacitidine in patients with advanced myelodysplastic syndrome (MDS). Am. Soc. Clin. Oncol. 2019. [Google Scholar] [CrossRef]
- Kizilbash, S.H.; McBrayer, S.; Port, J.; Reid, J.M.; Lanza, I.; Allred, J.B.; Chakravarti, A.; Kunos, C.; Adjei, A.A. A phase Ib trial of CB-839 (telaglenastat) in combination with radiation therapy and temozolomide in patients with IDH-mutated diffuse astrocytoma and anaplastic astrocytoma (NCT03528642). Am. Soc. Clin. Oncol. 2019. [Google Scholar] [CrossRef]
- Wang, E.S.; Frankfurt, O.; Orford, K.W.; Bennett, M.; Flinn, I.W.; Maris, M.; Konopleva, M. Phase 1 study of CB-839, a first-in-class, orally administered small molecule inhibitor of glutaminase in patients with relapsed/refractory leukemia. Blood 2015, 126, 2566. [Google Scholar] [CrossRef]
- Lee, G.K.; Park, H.J.; Macleod, M.; Chandler, P.; Munn, D.H.; Mellor, A.L. Tryptophan deprivation sensitizes activated T cells to apoptosis prior to cell division. Immunology 2002, 107, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Shafizadeh, E.; Attwood, J.T.; Bondarev, I.; Pashine, A.; Mellor, A.L. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J. Exp. Med. 1999, 189, 1363–1372. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Huang, L.; Bradley, J.; Liu, K.; Bardhan, K.; Ron, D.; Mellor, A.L.; Munn, D.H.; McGaha, T.L. GCN2-dependent metabolic stress is essential for endotoxemic cytokine induction and pathology. Mol. Cell. Biol. 2014, 34, 428–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bessede, A.; Gargaro, M.; Pallotta, M.T.; Matino, D.; Servillo, G.; Brunacci, C.; Bicciato, S.; Mazza, E.M.; Macchiarulo, A.; Vacca, C. Aryl hydrocarbon receptor control of a disease tolerance defence pathway. Nature 2014, 511, 184–190. [Google Scholar] [CrossRef] [Green Version]
- Uyttenhove, C.; Pilotte, L.; Théate, I.; Stroobant, V.; Colau, D.; Parmentier, N.; Boon, T.; Van den Eynde, B.J. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2, 3-dioxygenase. Nat. Med. 2003, 9, 1269–1274. [Google Scholar] [CrossRef]
- Okamoto, A.; Nikaido, T.; Ochiai, K.; Takakura, S.; Saito, M.; Aoki, Y.; Ishii, N.; Yanaihara, N.; Yamada, K.; Takikawa, O. Indoleamine 2, 3-dioxygenase serves as a marker of poor prognosis in gene expression profiles of serous ovarian cancer cells. Clin. Cancer Res. 2005, 11, 6030–6039. [Google Scholar] [CrossRef] [Green Version]
- Minhas, P.S.; Liu, L.; Moon, P.K.; Joshi, A.U.; Dove, C.; Mhatre, S.; Contrepois, K.; Wang, Q.; Lee, B.A.; Coronado, M. Macrophage de novo NAD+ synthesis specifies immune function in aging and inflammation. Nat. Immunol. 2019, 20, 50–63. [Google Scholar] [CrossRef]
- Munn, D.H.; Sharma, M.D.; Hou, D.; Baban, B.; Lee, J.R.; Antonia, S.J.; Messina, J.L.; Chandler, P.; Koni, P.A.; Mellor, A.L. Expression of indoleamine 2, 3-dioxygenase by plasmacytoid dendritic cells in tumor-draining lymph nodes. J. Clin. Investig. 2004, 114, 280–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinlich, G.; Murr, C.; Richardsen, L.; Winkler, C.; Fuchs, D. Decreased serum tryptophan concentration predicts poor prognosis in malignant melanoma patients. Dermatology 2007, 214, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Holmgaard, R.B.; Zamarin, D.; Munn, D.H.; Wolchok, J.D.; Allison, J.P. Indoleamine 2, 3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J. Exp. Med. 2013, 210, 1389–1402. [Google Scholar] [CrossRef] [PubMed]
- Cronin, S.J.; Seehus, C.; Weidinger, A.; Talbot, S.; Reissig, S.; Seifert, M.; Pierson, Y.; McNeill, E.; Longhi, M.S.; Turnes, B.L. The metabolite BH4 controls T cell proliferation in autoimmunity and cancer. Nature 2018, 563, 564–568. [Google Scholar] [CrossRef] [PubMed]
- Gu, T.; Rowswell-Turner, R.B.; Kilinc, M.O.; Egilmez, N.K. Central role of IFNγ–indoleamine 2, 3-dioxygenase axis in regulation of interleukin-12–mediated antitumor immunity. Cancer Res. 2010, 70, 129–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friberg, M.; Jennings, R.; Alsarraj, M.; Dessureault, S.; Cantor, A.; Extermann, M.; Mellor, A.L.; Munn, D.H.; Antonia, S.J. Indoleamine 2, 3-dioxygenase contributes to tumor cell evasion of T cell-mediated rejection. Int. J. Cancer 2002, 101, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Labadie, B.W.; Bao, R.; Luke, J.J. Reimagining IDO pathway inhibition in cancer immunotherapy via downstream focus on the tryptophan–kynurenine–aryl hydrocarbon axis. Clin. Cancer Res. 2019, 25, 1462–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M. L-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell 2016, 167, 829–842.e813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, P.C.; Zea, A.H.; Culotta, K.S.; Zabaleta, J.; Ochoa, J.B.; Ochoa, A.C. Regulation of t cell receptor cd3ζ chain expression byl-arginine. J. Biol. Chem. 2002, 277, 21123–21129. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, P.C.; Quiceno, D.G.; Ochoa, A.C. L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood 2007, 109, 1568–1573. [Google Scholar] [CrossRef] [Green Version]
- El-Gayar, S.; Thüring-Nahler, H.; Pfeilschifter, J.; Röllinghoff, M.; Bogdan, C. Translational control of inducible nitric oxide synthase by IL-13 and arginine availability in inflammatory macrophages. J. Immunol. 2003, 171, 4561–4568. [Google Scholar] [CrossRef] [Green Version]
- Pesce, J.T.; Ramalingam, T.R.; Mentink-Kane, M.M.; Wilson, M.S.; El Kasmi, K.C.; Smith, A.M.; Thompson, R.W.; Cheever, A.W.; Murray, P.J.; Wynn, T.A. Arginase-1–expressing macrophages suppress Th2 cytokine–driven inflammation and fibrosis. Plos Pathog. 2009, 5. [Google Scholar] [CrossRef] [Green Version]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cobbold, S.P.; Adams, E.; Farquhar, C.A.; Nolan, K.F.; Howie, D.; Lui, K.O.; Fairchild, P.J.; Mellor, A.L.; Ron, D.; Waldmann, H. Infectious tolerance via the consumption of essential amino acids and mTOR signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 12055–12060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doedens, A.L.; Stockmann, C.; Rubinstein, M.P.; Liao, D.; Zhang, N.; DeNardo, D.G.; Coussens, L.M.; Karin, M.; Goldrath, A.W.; Johnson, R.S. Macrophage expression of hypoxia-inducible factor-1α suppresses T-cell function and promotes tumor progression. Cancer Res. 2010, 70, 7465–7475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharda, D.R.; Yu, S.; Ray, M.; Squadrito, M.L.; De Palma, M.; Wynn, T.A.; Morris, S.M.; Hankey, P.A. Regulation of macrophage arginase expression and tumor growth by the Ron receptor tyrosine kinase. J. Immunol. 2011, 187, 2181–2192. [Google Scholar] [CrossRef]
- Rodriguez, P.C.; Quiceno, D.G.; Zabaleta, J.; Ortiz, B.; Zea, A.H.; Piazuelo, M.B.; Delgado, A.; Correa, P.; Brayer, J.; Sotomayor, E.M. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004, 64, 5839–5849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steggerda, S.M.; Bennett, M.K.; Chen, J.; Emberley, E.; Huang, T.; Janes, J.R.; Li, W.; MacKinnon, A.L.; Makkouk, A.; Marguier, G. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J. Immunother. Cancer 2017, 5, 101. [Google Scholar] [CrossRef]
- Papadopoulos, K.P.; Tsai, F.Y.-C.; Bauer, T.M.; Muigai, L.; Liang, Y.; Bennett, M.K.; Orford, K.W.; Fu, S. CX-1158-101: A first-in-human phase 1 study of CB-1158, a small molecule inhibitor of arginase, as monotherapy and in combination with an anti-PD-1 checkpoint inhibitor in patients (pts) with solid tumors. Am. Soc. Clin. Oncol. 2017. [Google Scholar] [CrossRef]
- Naing, A.; Bauer, T.; Papadopoulos, K.; Rahma, O.; Tsai, F.; Garralda, E.; Naidoo, J.; Pai, S.; Gibson, M.; Rybkin, I. Phase I study of the arginase inhibitor INCB001158 (1158) alone and in combination with pembrolizumab (PEM) in patients (Pts) with advanced/metastatic (adv/met) solid tumours. Ann. Oncol. 2019, 30, v160. [Google Scholar] [CrossRef]
- Hui, S.; Ghergurovich, J.M.; Morscher, R.J.; Jang, C.; Teng, X.; Lu, W.; Esparza, L.A.; Reya, T.; Zhan, L.; Guo, J.Y. Glucose feeds the TCA cycle via circulating lactate. Nature 2017, 551, 115–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faubert, B.; Li, K.Y.; Cai, L.; Hensley, C.T.; Kim, J.; Zacharias, L.G.; Yang, C.; Do, Q.N.; Doucette, S.; Burguete, D. Lactate metabolism in human lung tumors. Cell 2017, 171, 358–371.e359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ippolito, L.; Morandi, A.; Giannoni, E.; Chiarugi, P. Lactate: A metabolic driver in the tumour landscape. Trends Biochem. Sci. 2019, 44, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Husain, Z.; Huang, Y.; Seth, P.; Sukhatme, V.P. Tumor-derived lactate modifies antitumor immune response: Effect on myeloid-derived suppressor cells and NK cells. J. Immunol. 2013, 191, 1486–1495. [Google Scholar] [CrossRef]
- Angelin, A.; Gil-de-Gómez, L.; Dahiya, S.; Jiao, J.; Guo, L.; Levine, M.H.; Wang, Z.; Quinn III, W.J.; Kopinski, P.K.; Wang, L. Foxp3 reprograms T cell metabolism to function in low-glucose, high-lactate environments. Cell Metab. 2017, 25, 1282–1293.e1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K. LDHA-associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef] [Green Version]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S. Inhibitory effect of tumor cell–derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef]
- Huber, V.; Camisaschi, C.; Berzi, A.; Ferro, S.; Lugini, L.; Triulzi, T.; Tuccitto, A.; Tagliabue, E.; Castelli, C.; Rivoltini, L. Cancer acidity: An ultimate frontier of tumor immune escape and a novel target of immunomodulation. Semin. Cancer Biol. 2017, 43, 74–89. [Google Scholar] [CrossRef]
- Lacroix, R.; Rozeman, E.A.; Kreutz, M.; Renner, K.; Blank, C.U. Targeting tumor-associated acidity in cancer immunotherapy. Cancer Immunol. Immunother. 2018, 67, 1331–1348. [Google Scholar] [CrossRef]
- Peng, M.; Yin, N.; Chhangawala, S.; Xu, K.; Leslie, C.S.; Li, M.O. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science 2016, 354, 481–484. [Google Scholar] [CrossRef]
- Farabegoli, F.; Vettraino, M.; Manerba, M.; Fiume, L.; Roberti, M.; Di Stefano, G. Galloflavin, a new lactate dehydrogenase inhibitor, induces the death of human breast cancer cells with different glycolytic attitude by affecting distinct signaling pathways. Eur. J. Pharm. Sci. 2012, 47, 729–738. [Google Scholar] [CrossRef]
- Patel, C.H.; Leone, R.D.; Horton, M.R.; Powell, J.D. Targeting metabolism to regulate immune responses in autoimmunity and cancer. Nat. Rev. Drug Discov. 2019, 18, 669–688. [Google Scholar] [CrossRef]
- Miller, W.L.; Thomas, R.A.; Berne, R.M.; Rubio, R. Adenosine production in the ischemic kidney. Circ. Res. 1978, 43, 390–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorrentino, C.; Miele, L.; Porta, A.; Pinto, A.; Morello, S. Myeloid-derived suppressor cells contribute to A2B adenosine receptor-induced VEGF production and angiogenesis in a mouse melanoma model. Oncotarget 2015, 6, 27478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Huang, L.; Ye, H.; Song, S.P.; Bajwa, A.; Lee, S.J.; Moser, E.K.; Jaworska, K.; Kinsey, G.R.; Day, Y.J. Dendritic cells tolerized with adenosine A 2A R agonist attenuate acute kidney injury. J. Clin. Investig. 2012, 122, 3931–3942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vijayan, D.; Young, A.; Teng, M.W.; Smyth, M.J. Targeting immunosuppressive adenosine in cancer. Nat. Rev. Cancer 2017, 17, 709. [Google Scholar] [CrossRef]
- Cai, X.Y.; Wang, X.F.; Li, J.; Dong, J.N.; Liu, J.Q.; Li, N.P.; Yun, B.; Xia, R.L.; Qin, J.; Sun, Y.H. High expression of CD39 in gastric cancer reduces patient outcome following radical resection. Oncol. Lett. 2016, 12, 4080–4086. [Google Scholar] [CrossRef] [Green Version]
- Turcotte, M.; Spring, K.; Pommey, S.; Chouinard, G.; Cousineau, I.; George, J.; Chen, G.M.; Gendoo, D.M.; Haibe-Kains, B.; Karn, T. CD73 is associated with poor prognosis in high-grade serous ovarian cancer. Cancer Res. 2015, 75, 4494–4503. [Google Scholar] [CrossRef] [Green Version]
- Inoue, Y.; Yoshimura, K.; Kurabe, N.; Kahyo, T.; Kawase, A.; Tanahashi, M.; Ogawa, H.; Inui, N.; Funai, K.; Shinmura, K. Prognostic impact of CD73 and A2A adenosine receptor expression in non-small-cell lung cancer. Oncotarget 2017, 8, 8738. [Google Scholar] [CrossRef] [Green Version]
- Maj, T.; Wang, W.; Crespo, J.; Zhang, H.; Wang, W.; Wei, S.; Zhao, L.; Vatan, L.; Shao, I.; Szeliga, W. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat. Immunol. 2017, 18, 1332. [Google Scholar] [CrossRef]
- Sun, X.; Wu, Y.; Gao, W.; Enjyoji, K.; Csizmadia, E.; Müller, C.E.; Murakami, T.; Robson, S.C. CD39/ENTPD1 expression by CD4+ Foxp3+ regulatory T cells promotes hepatic metastatic tumor growth in mice. Gastroenterology 2010, 139, 1030–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.; Apasov, S.; Koshiba, M.; Sitkovsky, M. Role of A2a extracellular adenosine receptor-mediated signaling in adenosine-mediated inhibition of T-cell activation and expansion. Bloodthe J. Am. Soc. Hematol. 1997, 90, 1600–1610. [Google Scholar]
- Ohta, A.; Kini, R.; Ohta, A.; Subramanian, M.; Madasu, M.; Sitkovsky, M. The development and immunosuppressive functions of CD4+ CD25+ FoxP3+ regulatory T cells are under influence of the adenosine-A2A adenosine receptor pathway. Front. Immunol. 2012, 3, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allard, B.; Pommey, S.; Smyth, M.J.; Stagg, J. Targeting CD73 enhances the antitumor activity of anti-PD-1 and anti-CTLA-4 mAbs. Clin. Cancer Res. 2013, 19, 5626–5635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iannone, R.; Miele, L.; Maiolino, P.; Pinto, A.; Morello, S. Adenosine limits the therapeutic effectiveness of anti-CTLA4 mAb in a mouse melanoma model. Am. J. Cancer Res. 2014, 4, 172. [Google Scholar]
- Beavis, P.A.; Milenkovski, N.; Henderson, M.A.; John, L.B.; Allard, B.; Loi, S.; Kershaw, M.H.; Stagg, J.; Darcy, P.K. Adenosine receptor 2A blockade increases the efficacy of anti–PD-1 through enhanced antitumor T-cell responses. Cancer Immunol. Res. 2015, 3, 506–517. [Google Scholar] [CrossRef] [Green Version]
- Fong, L.; Forde, P.M.; Powderly, J.D.; Goldman, J.W.; Nemunaitis, J.J.; Luke, J.J.; Hellmann, M.D.; Kummar, S.; Doebele, R.C.; Mahadevan, D. Safety and clinical activity of adenosine A2a receptor (A2aR) antagonist, CPI-444, in anti-PD1/PDL1 treatment-refractory renal cell (RCC) and non-small cell lung cancer (NSCLC) patients. Am. Soc. Clin. Oncol. 2017. [Google Scholar] [CrossRef]
- Hammami, A.; Allard, D.; Allard, B.; Stagg, J. Targeting the adenosine pathway for cancer immunotherapy. Semin. Immunol. 2019, 42, 101304. [Google Scholar] [CrossRef]
- Parolini, I.; Federici, C.; Raggi, C.; Lugini, L.; Palleschi, S.; De Milito, A.; Coscia, C.; Iessi, E.; Logozzi, M.; Molinari, A. Microenvironmental pH is a key factor for exosome traffic in tumor cells. J. Biol. Chem. 2009, 284, 34211–34222. [Google Scholar] [CrossRef] [Green Version]
- Qu, J.-L.; Qu, X.-J.; Zhao, M.-F.; Teng, Y.-E.; Zhang, Y.; Hou, K.-Z.; Jiang, Y.-H.; Yang, X.-H.; Liu, Y.-P. Gastric cancer exosomes promote tumour cell proliferation through PI3K/Akt and MAPK/ERK activation. Dig. Liver Dis. 2009, 41, 875–880. [Google Scholar] [CrossRef]
- Zhao, H.; Yang, L.; Baddour, J.; Achreja, A.; Bernard, V.; Moss, T.; Marini, J.C.; Tudawe, T.; Seviour, E.G.; San Lucas, F.A. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. elife 2016, 5, e10250. [Google Scholar] [CrossRef] [PubMed]
- Achreja, A.; Zhao, H.; Yang, L.; Yun, T.H.; Marini, J.; Nagrath, D. Exo-MFA–a 13C metabolic flux analysis framework to dissect tumor microenvironment-secreted exosome contributions towards cancer cell metabolism. Metab. Eng. 2017, 43, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Lazar, I.; Clement, E.; Dauvillier, S.; Milhas, D.; Ducoux-Petit, M.; Legonidec, S.; Moro, C.; Soldan, V.; Dalle, S.; Balor, S. Adipocyte exosomes promote melanoma aggressiveness through fatty acid oxidation: A novel mechanism linking obesity and cancer. Cancer Res. 2016, 76, 4051–4057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015, 6, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patsoukis, N.; Brown, J.; Petkova, V.; Liu, F.; Li, L.; Boussiotis, V.A. Selective effects of PD-1 on Akt and Ras pathways regulate molecular components of the cell cycle and inhibit T cell proliferation. Sci. Signal. 2012, 5, ra46. [Google Scholar] [CrossRef] [Green Version]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell. Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Wenes, M.; Romero, P.; Huang, S.C.-C.; Fendt, S.-M.; Ho, P.-C. Navigating metabolic pathways to enhance antitumour immunity and immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 425–441. [Google Scholar] [CrossRef]
Figure 1.
Various gradients within the tumor microenvironment that are differentially associated with anti-tumor activity and tumor growth. Tumor cells are known for their aberrant metabolic activity that leads to local depletion of a variety of nutrients, including glucose, glutamine, arginine and tryptophan, which effectively hinder anti-tumor activities provided by immune cells that also depend on these nutrients. In addition, metabolites such as lactate, kynurenine and adenosine are released by tumor cells, dampening anti-tumor immunity along with hypoxia.
Figure 1.
Various gradients within the tumor microenvironment that are differentially associated with anti-tumor activity and tumor growth. Tumor cells are known for their aberrant metabolic activity that leads to local depletion of a variety of nutrients, including glucose, glutamine, arginine and tryptophan, which effectively hinder anti-tumor activities provided by immune cells that also depend on these nutrients. In addition, metabolites such as lactate, kynurenine and adenosine are released by tumor cells, dampening anti-tumor immunity along with hypoxia.


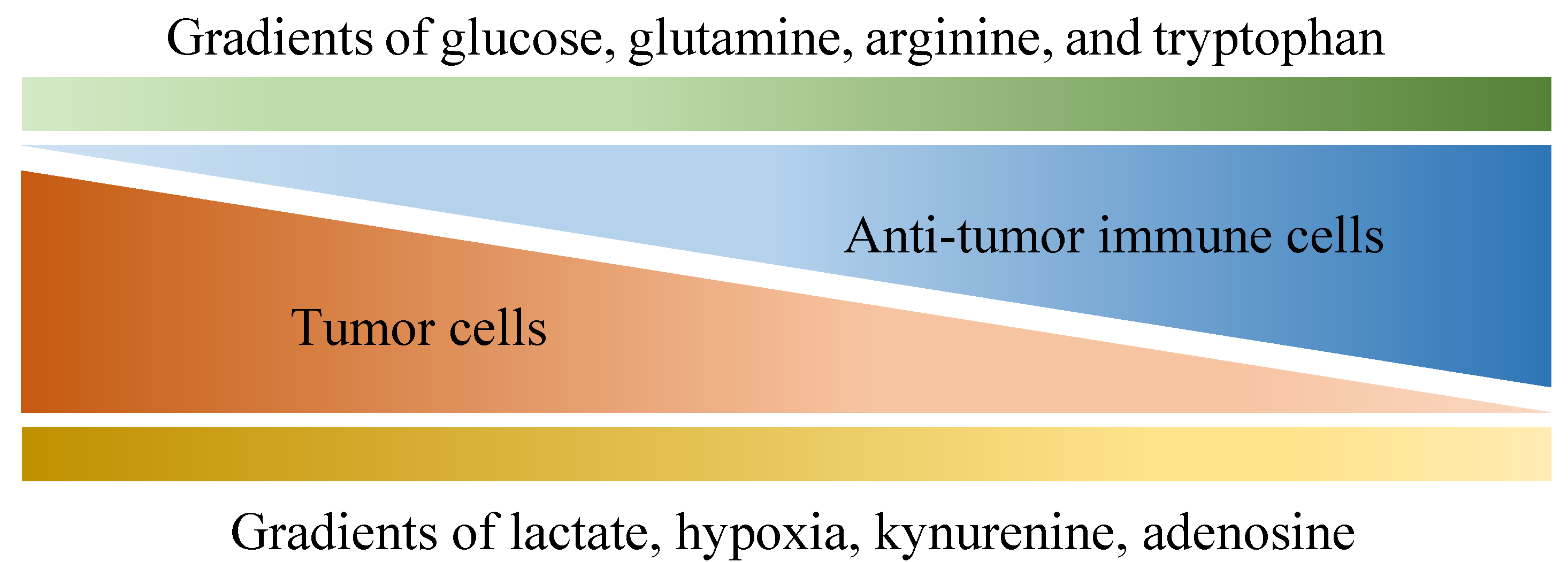{kind=link}
Table 1.
The role of different metabolic pathways in different kinds of T cells. Increased glycolysis and fatty acid synthesis is required for proliferation and differentiation of effector T cells upon activation. On the other hand, fatty acid oxidation is important for the development of CD8+ T cell memory and for the differentiation of CD4+ regulatory T cells as well. OXPHOS, oxidative phosphorylation; FAO, fatty acid oxidation; FAS, fatty acid synthesis.
Table 1.
The role of different metabolic pathways in different kinds of T cells. Increased glycolysis and fatty acid synthesis is required for proliferation and differentiation of effector T cells upon activation. On the other hand, fatty acid oxidation is important for the development of CD8+ T cell memory and for the differentiation of CD4+ regulatory T cells as well. OXPHOS, oxidative phosphorylation; FAO, fatty acid oxidation; FAS, fatty acid synthesis.
| Glycolysis | OXPHOS | FAO | FAS | |
|---|---|---|---|---|
| Naïve T cell | ○ | ○ | ||
| Effector T cell | ○ | ○ | ○ | |
| Memory T cell | ○ | ○ | ○ | |
| Regulatory T cell | ○ | ○ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Yang, H.-Y.; Wu, C.-Y.; Powell, J.D.; Lu, K.-L. Manipulation of Metabolic Pathways and Its Consequences for Anti-Tumor Immunity: A Clinical Perspective. Int. J. Mol. Sci. 2020, 21, 4030. https://doi.org/10.3390/ijms21114030
AMA Style
Yang H-Y, Wu C-Y, Powell JD, Lu K-L. Manipulation of Metabolic Pathways and Its Consequences for Anti-Tumor Immunity: A Clinical Perspective. International Journal of Molecular Sciences. 2020; 21(11):4030. https://doi.org/10.3390/ijms21114030
Chicago/Turabian StyleYang, Huang-Yu, Chao-Yi Wu, Jonathan D. Powell, and Kun-Lin Lu. 2020. "Manipulation of Metabolic Pathways and Its Consequences for Anti-Tumor Immunity: A Clinical Perspective" International Journal of Molecular Sciences 21, no. 11: 4030. https://doi.org/10.3390/ijms21114030
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.