Anti-Citrullinated Protein Antibodies in Patients with Rheumatoid Arthritis: Biological Effects and Mechanisms of Immunopathogenesis


Abstract
:1. Introduction: Overview of ACPAs in Patients with Rheumatoid Arthritis
2. Characteristics of ACPAs in RA
2.1. ACPA Isotypes
2.2. ACPA Specificity and Avidity
2.3. N-Linked Glycosylation of ACPA
2.4. Synergism between ACPAs and RF through Immune Complex Formation
3. Immunopathogenesis of ACPAs in RA
3.1. Dysregulated Citrullination Fuels the Production of Citrullinated Neoantigens for ACPA Targeting
3.1.1. Environmental Factors and ACPA Production
3.1.2. Cellular Death Pathways and ACPA Production
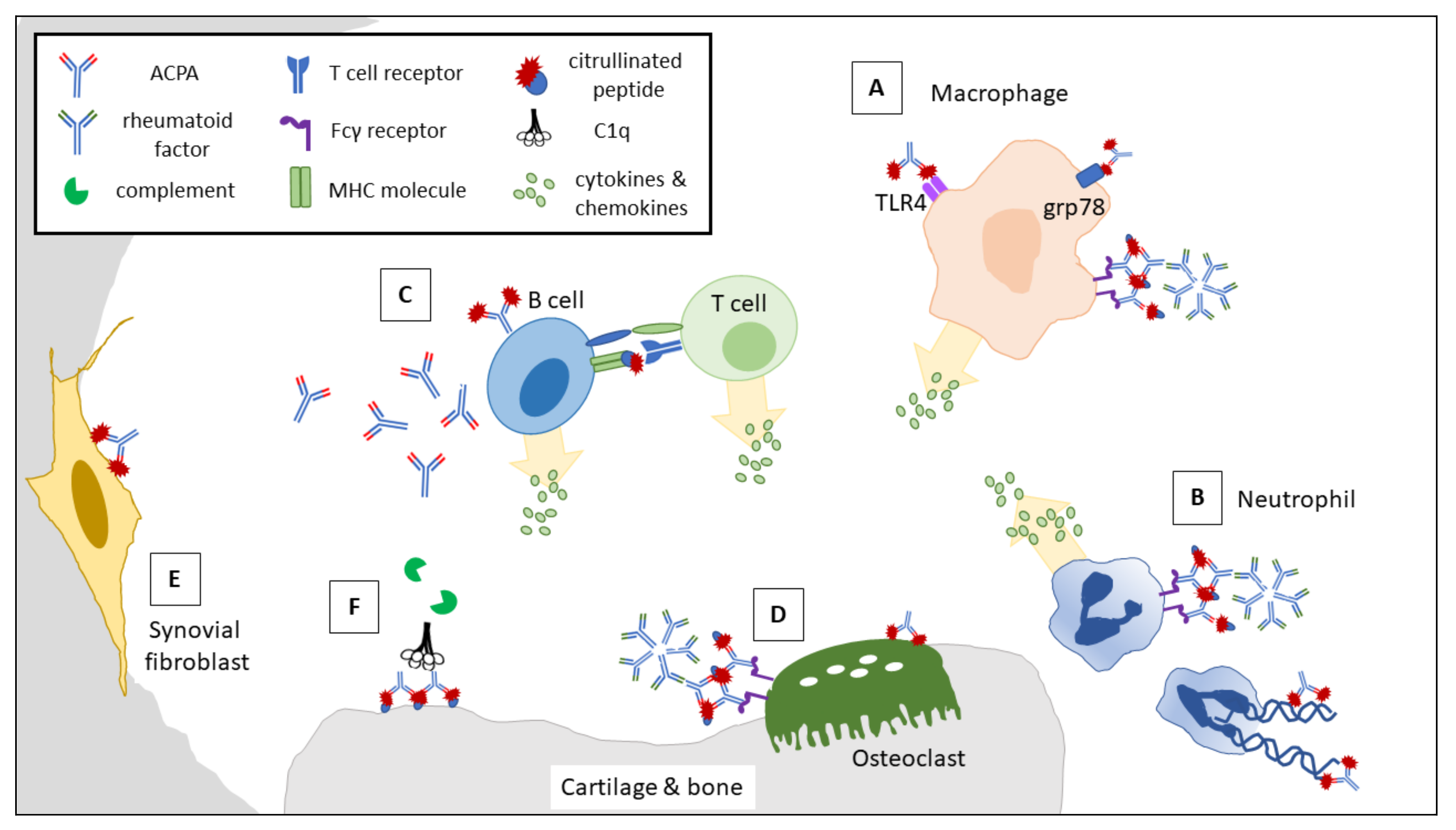
3.2. Interactions between ACPAs and RA-Related Immune Cells and Mediators
3.2.1. ACPAs Activate Macrophages via IC Formation and Agonistic Activity to Elicit Proinflammatory Cytokine Production
3.2.2. ACPA-Forming ICs Promote NETosis and the Release of Degradative Enzymes and Reactive Oxygen Species upon Binding with FcγR
3.2.3. ACPAs Activate Complement via the Classical and Alternative Pathways
3.2.4. Autoreactive B Cells Baring Surface ACPAs Promote T Cell Differentiation and Secrete Proinflammatory Cytokines
3.3. Interactions between ACPAs and Synovial Residential Cells
3.3.1. Binding of ACPAs to form ICs via FcγR Promotes Osteoclast Differentiation and Proinflammatory Cytokine Production
3.3.2. ACPAs Interact with Cellular Citrullinated Antigens and Promote Synovial Fibroblast Migration
3.3.3. ACPAs Induce Joint Destruction by Cross-Reacting with Type II Collagen Fibers
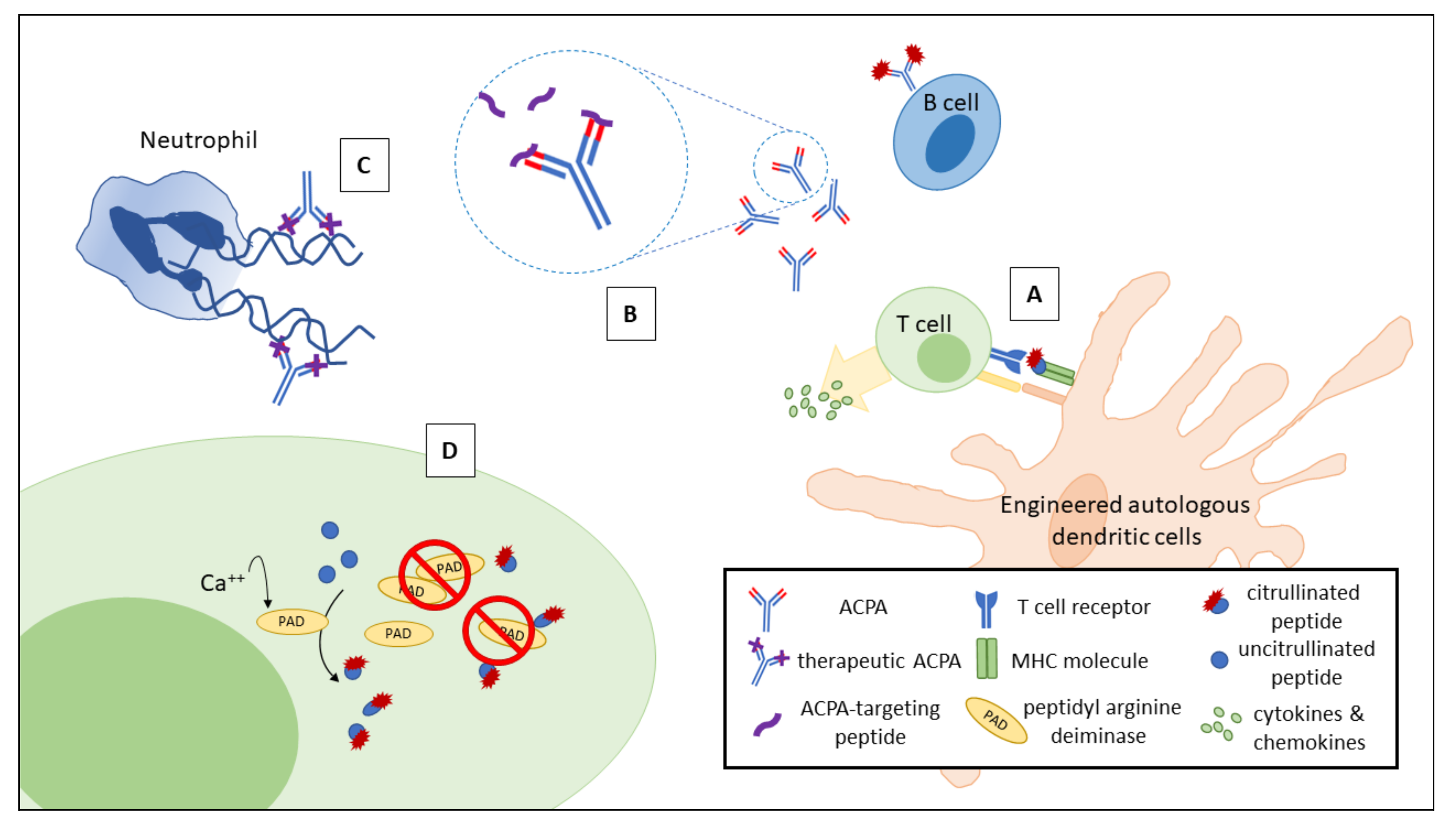
4. Potential of Therapeutic Approaches Targeting ACPAs for Patients with RA
5. Conclusions
Funding
Conflicts of Interest
Abbreviations
| Aa | Aggregatibacter actinomycetemcomitans |
| ACPA | anti-citrullinated protein antibody |
| APCs | antigen presenting cells |
| C5a | complement 5a |
| CCPs | cyclic citrullinated peptides |
| ERK | extracellular signal-regulated kinases |
| Fab | antigen-binding fragment |
| Fc | crystallizable fragment |
| FcγR | Fc gamma receptor |
| HLA | human leukocyte antigen |
| IC | immune complex |
| IFNγ | interferon-gamma |
| IL | Interleukin |
| JNK | c-Jun N-terminal kinase |
| MAC | membrane attack complex |
| MMPs | matrix metalloproteinases |
| NET | neutrophil extracellular trap |
| PAD | peptidyl arginine deiminase |
| P. gingivalis | Porphyromomas gingivalis |
| RA | rheumatoid arthritis |
| RF | rheumatoid factor |
| ROS | reactive oxygen species |
| SE | shared epitope |
| M-CSF | macrophage colony-stimulating factor |
| NF-κB | nuclear factor-kappaB |
| RANKL | receptor activator of nuclear factor kappa-Β ligand |
| TLR | Toll-like receptor |
| TNF-α | tumor necrosis factor-alpha |
References
- McInnes, I.B.; Schett, G. The pathogenesis of rheumatoid arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aletaha, D.; Neogi, T.; Silman, A.J.; Funovits, J.; Felson, D.T.; Bingham, C.O., III; Birnbaum, N.S.; Burmester, G.R.; Bykerk, V.P.; Cohen, M.D.; et al. 2010 Rheumatoid arthritis classification criteria: An American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010, 62, 2569–2581. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, G.C.; Scheinberg, M.A.; Aparecida da Silva, M.; Maciel, S. Anti-cyclic citrullinated peptide antibodies in advanced rheumatoid arthritis. Ann. Intern. Med. 2003, 139, 234–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.M.; Schur, P.H. Clinical utility of the anti-CCP assay in patients with rheumatic diseases. Ann. Rheum. Dis. 2003, 62, 870–874. [Google Scholar] [CrossRef] [Green Version]
- Van Zanten, A.; Arends, S.; Roozendaal, C.; Limburg, P.C.; Maas, F.; Trouw, L.A.; Toes, R.E.M.; Huizinga, T.W.J.; Bootsma, H.; Brouwer, E. Presence of anticitrullinated protein antibodies in a large population-based cohort from the Netherlands. Ann. Rheum. Dis. 2017, 76, 1184–1190. [Google Scholar] [CrossRef]
- Van Beers, J.J.; Willemze, A.; Jansen, J.J.; Engbers, G.H.; Salden, M.; Raats, J.; Drijfhout, J.W.; van der Helm-van Mil, A.H.; Toes, R.E.; Pruijn, G.J. ACPA fine-specificity profiles in early rheumatoid arthritis patients do not correlate with clinical features at baseline or with disease progression. Arthritis Res. Ther. 2013, 15, R140. [Google Scholar] [CrossRef] [Green Version]
- Schwenzer, A.; Jiang, X.; Mikuls, T.R.; Payne, J.B.; Sayles, H.R.; Quirke, A.M.; Kessler, B.M.; Fischer, R.; Venables, P.J.; Lundberg, K.; et al. Identification of an immunodominant peptide from citrullinated tenascin-C as a major target for autoantibodies in rheumatoid arthritis. Ann. Rheum. Dis. 2016, 75, 1876–1883. [Google Scholar] [CrossRef] [Green Version]
- Johansson, L.; Pratesi, F.; Brink, M.; Arlestig, L.; D’Amato, C.; Bartaloni, D.; Migliorini, P.; Rantapaa-Dahlqvist, S. Antibodies directed against endogenous and exogenous citrullinated antigens pre-date the onset of rheumatoid arthritis. Arthritis Res. Ther. 2016, 18, 127. [Google Scholar] [CrossRef] [Green Version]
- Vallbracht, I.; Rieber, J.; Oppermann, M.; Forger, F.; Siebert, U.; Helmke, K. Diagnostic and clinical value of anti-cyclic citrullinated peptide antibodies compared with rheumatoid factor isotypes in rheumatoid arthritis. Ann. Rheum. Dis. 2004, 63, 1079–1084. [Google Scholar] [CrossRef] [Green Version]
- Visser, H.; le Cessie, S.; Vos, K.; Breedveld, F.C.; Hazes, J.M. How to diagnose rheumatoid arthritis early: A prediction model for persistent (erosive) arthritis. Arthritis Rheum. 2002, 46, 357–365. [Google Scholar] [CrossRef]
- Kroot, E.J.; de Jong, B.A.; van Leeuwen, M.A.; Swinkels, H.; van den Hoogen, F.H.; van’t Hof, M.; van de Putte, L.B.; van Rijswijk, M.H.; van Venrooij, W.J.; van Riel, P.L. The prognostic value of anti-cyclic citrullinated peptide antibody in patients with recent-onset rheumatoid arthritis. Arthritis Rheum. 2000, 43, 1831–1835. [Google Scholar] [CrossRef]
- Nielen, M.M.; van Schaardenburg, D.; Reesink, H.W.; van de Stadt, R.J.; van der Horst-Bruinsma, I.E.; de Koning, M.H.; Habibuw, M.R.; Vandenbroucke, J.P.; Dijkmans, B.A. Specific autoantibodies precede the symptoms of rheumatoid arthritis: A study of serial measurements in blood donors. Arthritis Rheum. 2004, 50, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Koga, T.; Okada, A.; Fukuda, T.; Hidaka, T.; Ishii, T.; Ueki, Y.; Kodera, T.; Nakashima, M.; Takahashi, Y.; Honda, S.; et al. Anti-citrullinated peptide antibodies are the strongest predictor of clinically relevant radiographic progression in rheumatoid arthritis patients achieving remission or low disease activity: A post hoc analysis of a nationwide cohort in Japan. PLoS ONE 2017, 12, e0175281. [Google Scholar] [CrossRef] [PubMed]
- Van den Broek, M.; Dirven, L.; Klarenbeek, N.B.; Molenaar, T.H.; Han, K.H.; Kerstens, P.J.; Huizinga, T.W.; Dijkmans, B.A.; Allaart, C.F. The association of treatment response and joint damage with ACPA-status in recent-onset RA: A subanalysis of the 8-year follow-up of the BeSt study. Ann. Rheum. Dis. 2012, 71, 245–248. [Google Scholar] [CrossRef] [Green Version]
- Ajeganova, S.; Andersson, M.L.; Frostegard, J.; Hafstrom, I. Disease factors in early rheumatoid arthritis are associated with differential risks for cardiovascular events and mortality depending on age at onset: A 10-year observational cohort study. J. Rheumatol. 2013, 40, 1958–1966. [Google Scholar] [CrossRef]
- Reynisdottir, G.; Karimi, R.; Joshua, V.; Olsen, H.; Hensvold, A.H.; Harju, A.; Engstrom, M.; Grunewald, J.; Nyren, S.; Eklund, A.; et al. Structural changes and antibody enrichment in the lungs are early features of anti-citrullinated protein antibody-positive rheumatoid arthritis. Arthritis Rheumatol. 2014, 66, 31–39. [Google Scholar] [CrossRef]
- Sellam, J.; Hendel-Chavez, H.; Rouanet, S.; Abbed, K.; Combe, B.; Le Loet, X.; Tebib, J.; Sibilia, J.; Taoufik, Y.; Dougados, M.; et al. B cell activation biomarkers as predictive factors for the response to rituximab in rheumatoid arthritis: A six-month, national, multicenter, open-label study. Arthritis Rheum. 2011, 63, 933–938. [Google Scholar] [CrossRef]
- Isaacs, J.D.; Cohen, S.B.; Emery, P.; Tak, P.P.; Wang, J.; Lei, G.; Williams, S.; Lal, P.; Read, S.J. Effect of baseline rheumatoid factor and anticitrullinated peptide antibody serotype on rituximab clinical response: A meta-analysis. Ann. Rheum. Dis. 2013, 72, 329–336. [Google Scholar] [CrossRef]
- Chatzidionysiou, K.; Lie, E.; Nasonov, E.; Lukina, G.; Hetland, M.L.; Tarp, U.; Gabay, C.; van Riel, P.L.; Nordstrom, D.C.; Gomez-Reino, J.; et al. Highest clinical effectiveness of rituximab in autoantibody-positive patients with rheumatoid arthritis and in those for whom no more than one previous TNF antagonist has failed: Pooled data from 10 European registries. Ann. Rheum. Dis. 2011, 70, 1575–1580. [Google Scholar] [CrossRef]
- Ally, M.M.; Hodkinson, B.; Meyer, P.W.; Musenge, E.; Tintinger, G.R.; Tikly, M.; Anderson, R. Circulating anti-citrullinated peptide antibodies, cytokines and genotype as biomarkers of response to disease-modifying antirheumatic drug therapy in early rheumatoid arthritis. BMC Musculoskelet. Disord. 2015, 16, 130. [Google Scholar] [CrossRef] [Green Version]
- Lv, Q.; Yin, Y.; Li, X.; Shan, G.; Wu, X.; Liang, D.; Li, Y.; Zhang, X. The status of rheumatoid factor and anti-cyclic citrullinated peptide antibody are not associated with the effect of anti-TNFalpha agent treatment in patients with rheumatoid arthritis: A meta-analysis. PLoS ONE 2014, 9, e89442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pruijn, G.J.; Wiik, A.; van Venrooij, W.J. The use of citrullinated peptides and proteins for the diagnosis of rheumatoid arthritis. Arthritis Res. Ther. 2010, 12, 203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valesini, G.; Gerardi, M.C.; Iannuccelli, C.; Pacucci, V.A.; Pendolino, M.; Shoenfeld, Y. Citrullination and autoimmunity. Autoimmun. Rev. 2015, 14, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Holers, V.M.; Demoruelle, M.K.; Kuhn, K.A.; Buckner, J.H.; Robinson, W.H.; Okamoto, Y.; Norris, J.M.; Deane, K.D. Rheumatoid arthritis and the mucosal origins hypothesis: Protection turns to destruction. Nat. Rev. Rheumatol. 2018, 14, 542–557. [Google Scholar] [CrossRef] [PubMed]
- Malmstrom, V.; Catrina, A.I.; Klareskog, L. The immunopathogenesis of seropositive rheumatoid arthritis: From triggering to targeting. Nat. Rev. Immunol. 2017, 17, 60–75. [Google Scholar] [CrossRef]
- Makrygiannakis, D.; Hermansson, M.; Ulfgren, A.K.; Nicholas, A.P.; Zendman, A.J.; Eklund, A.; Grunewald, J.; Skold, C.M.; Klareskog, L.; Catrina, A.I. Smoking increases peptidylarginine deiminase 2 enzyme expression in human lungs and increases citrullination in BAL cells. Ann. Rheum. Dis. 2008, 67, 1488–1492. [Google Scholar] [CrossRef]
- Konig, M.F.; Andrade, F. A Critical Reappraisal of Neutrophil Extracellular Traps and NETosis Mimics Based on Differential Requirements for Protein Citrullination. Front. Immunol. 2016, 7, 461. [Google Scholar] [CrossRef] [Green Version]
- Konig, M.F.; Abusleme, L.; Reinholdt, J.; Palmer, R.J.; Teles, R.P.; Sampson, K.; Rosen, A.; Nigrovic, P.A.; Sokolove, J.; Giles, J.T.; et al. Aggregatibacter actinomycetemcomitans-induced hypercitrullination links periodontal infection to autoimmunity in rheumatoid arthritis. Sci. Transl. Med. 2016, 8, 369ra176. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, A.; Jantsch, V.; Khan, R.; Hartung, W.; Fischer, R.; Jantsch, J.; Ehrenstein, B.; Konig, M.F.; Andrade, F. Rheumatoid Arthritis-Associated Autoimmunity Due to Aggregatibacter actinomycetemcomitans and Its Resolution with Antibiotic Therapy. Front. Immunol. 2018, 9, 2352. [Google Scholar] [CrossRef] [Green Version]
- Volkov, M.; van Schie, K.A.; van der Woude, D. Autoantibodies and B Cells: The ABC of rheumatoid arthritis pathophysiology. Immunol. Rev. 2020, 294, 148–163. [Google Scholar] [CrossRef] [Green Version]
- Malmstrom, V.; Gronwall, C. The parallel worlds of ACPA-positive and RF-positive B cells. Nat. Rev. Rheumatol. 2018, 14, 626–628. [Google Scholar] [CrossRef] [PubMed]
- Sokolove, J.; Zhao, X.; Chandra, P.E.; Robinson, W.H. Immune complexes containing citrullinated fibrinogen costimulate macrophages via Toll-like receptor 4 and Fcgamma receptor. Arthritis Rheum. 2011, 63, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Trouw, L.A.; Haisma, E.M.; Levarht, E.W.; van der Woude, D.; Ioan-Facsinay, A.; Daha, M.R.; Huizinga, T.W.; Toes, R.E. Anti-cyclic citrullinated peptide antibodies from rheumatoid arthritis patients activate complement via both the classical and alternative pathways. Arthritis Rheum. 2009, 60, 1923–1931. [Google Scholar] [CrossRef] [PubMed]
- Rombouts, Y.; Ewing, E.; van de Stadt, L.A.; Selman, M.H.; Trouw, L.A.; Deelder, A.M.; Huizinga, T.W.; Wuhrer, M.; van Schaardenburg, D.; Toes, R.E.; et al. Anti-citrullinated protein antibodies acquire a pro-inflammatory Fc glycosylation phenotype prior to the onset of rheumatoid arthritis. Ann. Rheum. Dis 2015, 74, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Arlestig, L.; Mullazehi, M.; Kokkonen, H.; Rocklov, J.; Ronnelid, J.; Dahlqvist, S.R. Antibodies against cyclic citrullinated peptides of IgG, IgA and IgM isotype and rheumatoid factor of IgM and IgA isotype are increased in unaffected members of multicase rheumatoid arthritis families from northern Sweden. Ann. Rheum. Dis. 2012, 71, 825–829. [Google Scholar] [CrossRef] [Green Version]
- Engelmann, R.; Brandt, J.; Eggert, M.; Karberg, K.; Krause, A.; Neeck, G.; Mueller-Hilke, B. IgG1 and IgG4 are the predominant subclasses among auto-antibodies against two citrullinated antigens in RA. Rheumatology (Oxford) 2008, 47, 1489–1492. [Google Scholar] [CrossRef] [Green Version]
- Willis, V.C.; Demoruelle, M.K.; Derber, L.A.; Chartier-Logan, C.J.; Parish, M.C.; Pedraza, I.F.; Weisman, M.H.; Norris, J.M.; Holers, V.M.; Deane, K.D. Sputum autoantibodies in patients with established rheumatoid arthritis and subjects at risk of future clinically apparent disease. Arthritis Rheum. 2013, 65, 2545–2554. [Google Scholar] [CrossRef]
- Kokkonen, H.; Mullazehi, M.; Berglin, E.; Hallmans, G.; Wadell, G.; Ronnelid, J.; Rantapaa-Dahlqvist, S. Antibodies of IgG, IgA and IgM isotypes against cyclic citrullinated peptide precede the development of rheumatoid arthritis. Arthritis Res. Ther. 2011, 13, R13. [Google Scholar] [CrossRef] [Green Version]
- Demoruelle, M.K.; Harrall, K.K.; Ho, L.; Purmalek, M.M.; Seto, N.L.; Rothfuss, H.M.; Weisman, M.H.; Solomon, J.J.; Fischer, A.; Okamoto, Y.; et al. Anti-Citrullinated Protein Antibodies Are Associated With Neutrophil Extracellular Traps in the Sputum in Relatives of Rheumatoid Arthritis Patients. Arthritis Rheum. 2017, 69, 1165–1175. [Google Scholar] [CrossRef]
- Kinslow, J.D.; Blum, L.K.; Deane, K.D.; Demoruelle, M.K.; Okamoto, Y.; Parish, M.C.; Kongpachith, S.; Lahey, L.J.; Norris, J.M.; Robinson, W.H.; et al. Elevated IgA Plasmablast Levels in Subjects at Risk of Developing Rheumatoid Arthritis. Arthritis Rheum. 2016, 68, 2372–2383. [Google Scholar] [CrossRef] [Green Version]
- Van der Steen, L.P.; Bakema, J.E.; Sesarman, A.; Florea, F.; Tuk, C.W.; Kirtschig, G.; Hage, J.J.; Sitaru, C.; van Egmond, M. Blocking Fcalpha receptor I on granulocytes prevents tissue damage induced by IgA autoantibodies. J. Immunol. 2012, 189, 1594–1601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Venrooij, W.J.; Pruijn, G.J. Citrullination: A small change for a protein with great consequences for rheumatoid arthritis. Arthritis Res. 2000, 2, 249–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuhrmann, J.; Clancy, K.W.; Thompson, P.R. Chemical biology of protein arginine modifications in epigenetic regulation. Chem. Rev. 2015, 115, 5413–5461. [Google Scholar] [CrossRef] [Green Version]
- Van Beers, J.J.; Schwarte, C.M.; Stammen-Vogelzangs, J.; Oosterink, E.; Bozic, B.; Pruijn, G.J. The rheumatoid arthritis synovial fluid citrullinome reveals novel citrullinated epitopes in apolipoprotein E, myeloid nuclear differentiation antigen, and beta-actin. Arthritis Rheum. 2013, 65, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Y.; Wang, D.; Wilhelm, M.; Zolg, D.P.; Schmidt, T.; Schnatbaum, K.; Reimer, U.; Ponten, F.; Uhlen, M.; Hahne, H.; et al. Mining the Human Tissue Proteome for Protein Citrullination. Mol. Cell Proteom. 2018, 17, 1378–1391. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Chen, F.F.; Gao, W.B.; Wang, H.Y.; Zhao, N.W.; Xu, M.; Gao, D.Y.; Yu, W.; Yan, X.L.; Zhao, J.N.; et al. Identification of citrullinated peptides in the synovial fluid of patients with rheumatoid arthritis using LC-MALDI-TOF/TOF. Clin. Rheumatol. 2016, 35, 2185–2194. [Google Scholar] [CrossRef] [Green Version]
- Tilvawala, R.; Nguyen, S.H.; Maurais, A.J.; Nemmara, V.V.; Nagar, M.; Salinger, A.J.; Nagpal, S.; Weerapana, E.; Thompson, P.R. The Rheumatoid Arthritis-Associated Citrullinome. Cell Chem. Biol. 2018, 25, 691–770. [Google Scholar] [CrossRef] [Green Version]
- Romero, V.; Fert-Bober, J.; Nigrovic, P.A.; Darrah, E.; Haque, U.J.; Lee, D.M.; van Eyk, J.; Rosen, A.; Andrade, F. Immune-mediated pore-forming pathways induce cellular hypercitrullination and generate citrullinated autoantigens in rheumatoid arthritis. Sci. Transl. Med. 2013, 5, 209ra150. [Google Scholar] [CrossRef] [Green Version]
- Cambridge, G.; Leandro, M.J.; Lahey, L.J.; Fairhead, T.; Robinson, W.H.; Sokolove, J. B cell depletion with rituximab in patients with rheumatoid arthritis: Multiplex bead array reveals the kinetics of IgG and IgA antibodies to citrullinated antigens. J. Autoimmun. 2016, 70, 22–30. [Google Scholar] [CrossRef]
- Kampstra, A.S.B.; Dekkers, J.S.; Volkov, M.; Dorjee, A.L.; Hafkenscheid, L.; Kempers, A.C.; van Delft, M.; Kissel, T.; Reijm, S.; Janssen, G.M.C.; et al. Different classes of anti-modified protein antibodies are induced on exposure to antigens expressing only one type of modification. Ann. Rheum. Dis. 2019, 78, 908–916. [Google Scholar] [CrossRef]
- Shi, J.; Willemze, A.; Janssen, G.M.; van Veelen, P.A.; Drijfhout, J.W.; Cerami, A.; Huizinga, T.W.; Trouw, L.A.; Toes, R.E. Recognition of citrullinated and carbamylated proteins by human antibodies: Specificity, cross-reactivity and the ’AMC-Senshu’ method. Ann. Rheum. Dis. 2013, 72, 148–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, C.; Holmdahl, R. The structure, specificity and function of anti-citrullinated protein antibodies. Nat. Rev. Rheumatol. 2019, 15, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Ge, C.; Xu, B.; Liang, B.; Lonnblom, E.; Lundstrom, S.L.; Zubarev, R.A.; Ayoglu, B.; Nilsson, P.; Skogh, T.; Kastbom, A.; et al. Structural Basis of Cross-Reactivity of Anti-Citrullinated Protein Antibodies. Arthritis Rheumatol. 2019, 71, 210–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, C.; Tong, D.; Liang, B.; Lonnblom, E.; Schneider, N.; Hagert, C.; Viljanen, J.; Ayoglu, B.; Stawikowska, R.; Nilsson, P.; et al. Anti-citrullinated protein antibodies cause arthritis by cross-reactivity to joint cartilage. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Woude, D.; Rantapaa-Dahlqvist, S.; Ioan-Facsinay, A.; Onnekink, C.; Schwarte, C.M.; Verpoort, K.N.; Drijfhout, J.W.; Huizinga, T.W.; Toes, R.E.; Pruijn, G.J. Epitope spreading of the anti-citrullinated protein antibody response occurs before disease onset and is associated with the disease course of early arthritis. Ann. Rheum. Dis. 2010, 69, 1554–1561. [Google Scholar] [CrossRef] [PubMed]
- Kongpachith, S.; Lingampalli, N.; Ju, C.H.; Blum, L.K.; Lu, D.R.; Elliott, S.E.; Mao, R.; Robinson, W.H. Affinity Maturation of the Anti-Citrullinated Protein Antibody Paratope Drives Epitope Spreading and Polyreactivity in Rheumatoid Arthritis. Arthritis Rheumatol. 2019, 71, 507–517. [Google Scholar] [CrossRef]
- Titcombe, P.J.; Wigerblad, G.; Sippl, N.; Zhang, N.; Shmagel, A.K.; Sahlstrom, P.; Zhang, Y.; Barsness, L.O.; Ghodke-Puranik, Y.; Baharpoor, A.; et al. Pathogenic Citrulline-Multispecific B Cell Receptor Clades in Rheumatoid Arthritis. Arthritis Rheumatol. 2018, 70, 1933–1945. [Google Scholar] [CrossRef] [Green Version]
- Elliott, S.E.; Kongpachith, S.; Lingampalli, N.; Adamska, J.Z.; Cannon, B.J.; Mao, R.; Blum, L.K.; Robinson, W.H. Affinity Maturation Drives Epitope Spreading and Generation of Proinflammatory Anti-Citrullinated Protein Antibodies in Rheumatoid Arthritis. Arthritis Rheumatol. 2018, 70, 1946–1958. [Google Scholar] [CrossRef] [Green Version]
- Steen, J.; Forsstrom, B.; Sahlstrom, P.; Odowd, V.; Israelsson, L.; Krishnamurthy, A.; Badreh, S.; Mathsson Alm, L.; Compson, J.; Ramskold, D.; et al. Recognition of Amino Acid Motifs, Rather Than Specific Proteins, by Human Plasma Cell-Derived Monoclonal Antibodies to Posttranslationally Modified Proteins in Rheumatoid Arthritis. Arthritis Rheumatol. 2019, 71, 196–209. [Google Scholar] [CrossRef]
- Lu, D.R.; McDavid, A.N.; Kongpachith, S.; Lingampalli, N.; Glanville, J.; Ju, C.H.; Gottardo, R.; Robinson, W.H. T Cell-Dependent Affinity Maturation and Innate Immune Pathways Differentially Drive Autoreactive B Cell Responses in Rheumatoid Arthritis. Arthritis Rheumatol. 2018, 70, 1732–1744. [Google Scholar] [CrossRef] [Green Version]
- Imperiali, B.; O’Connor, S.E. Effect of N-linked glycosylation on glycopeptide and glycoprotein structure. Curr. Opin. Chem. Biol. 1999, 3, 643–649. [Google Scholar] [CrossRef]
- Anumula, K.R. Quantitative glycan profiling of normal human plasma derived immunoglobulin and its fragments Fab and Fc. J. Immunol. Methods 2012, 382, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Vergroesen, R.D.; Slot, L.M.; Hafkenscheid, L.; Koning, M.T.; van der Voort, E.I.H.; Grooff, C.A.; Zervakis, G.; Veelken, H.; Huizinga, T.W.J.; Rispens, T.; et al. B-cell receptor sequencing of anti-citrullinated protein antibody (ACPA) IgG-expressing B cells indicates a selective advantage for the introduction of N-glycosylation sites during somatic hypermutation. Ann. Rheum. Dis. 2018, 77, 956–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hafkenscheid, L.; Bondt, A.; Scherer, H.U.; Huizinga, T.W.; Wuhrer, M.; Toes, R.E.; Rombouts, Y. Structural Analysis of Variable Domain Glycosylation of Anti-Citrullinated Protein Antibodies in Rheumatoid Arthritis Reveals the Presence of Highly Sialylated Glycans. Mol. Cell Proteom. 2017, 16, 278–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hafkenscheid, L.; de Moel, E.; Smolik, I.; Tanner, S.; Meng, X.; Jansen, B.C.; Bondt, A.; Wuhrer, M.; Huizinga, T.W.J.; Toes, R.E.M.; et al. N-Linked Glycans in the Variable Domain of IgG Anti-Citrullinated Protein Antibodies Predict the Development of Rheumatoid Arthritis. Arthritis Rheumatol. 2019, 71, 1626–1633. [Google Scholar] [CrossRef] [Green Version]
- Rombouts, Y.; Willemze, A.; van Beers, J.J.; Shi, J.; Kerkman, P.F.; van Toorn, L.; Janssen, G.M.; Zaldumbide, A.; Hoeben, R.C.; Pruijn, G.J.; et al. Extensive glycosylation of ACPA-IgG variable domains modulates binding to citrullinated antigens in rheumatoid arthritis. Ann. Rheum. Dis. 2016, 75, 578–585. [Google Scholar] [CrossRef]
- Ohmi, Y.; Ise, W.; Harazono, A.; Takakura, D.; Fukuyama, H.; Baba, Y.; Narazaki, M.; Shoda, H.; Takahashi, N.; Ohkawa, Y.; et al. Sialylation converts arthritogenic IgG into inhibitors of collagen-induced arthritis. Nat. Commun. 2016, 7, 11205. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, K.A.; Steen, J.; Amara, K.; Titcombe, P.J.; Israelsson, L.; Lundstrom, S.L.; Zhou, D.; Zubarev, R.A.; Reed, E.; Piccoli, L.; et al. Variable domain N-linked glycosylation and negative surface charge are key features of monoclonal ACPA: Implications for B-cell selection. Eur J. Immunol. 2018, 48, 1030–1045. [Google Scholar] [CrossRef] [Green Version]
- Scherer, H.U.; van der Woude, D.; Ioan-Facsinay, A.; el Bannoudi, H.; Trouw, L.A.; Wang, J.; Haupl, T.; Burmester, G.R.; Deelder, A.M.; Huizinga, T.W.; et al. Glycan profiling of anti-citrullinated protein antibodies isolated from human serum and synovial fluid. Arthritis Rheum. 2010, 62, 1620–1629. [Google Scholar] [CrossRef]
- Kaneko, Y.; Nimmerjahn, F.; Ravetch, J.V. Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science 2006, 313, 670–673. [Google Scholar] [CrossRef] [Green Version]
- Rademacher, T.W.; Williams, P.; Dwek, R.A. Agalactosyl glycoforms of IgG autoantibodies are pathogenic. Proc. Natl. Acad. Sci. USA 1994, 91, 6123–6127. [Google Scholar] [CrossRef] [Green Version]
- Anquetil, F.; Clavel, C.; Offer, G.; Serre, G.; Sebbag, M. IgM and IgA rheumatoid factors purified from rheumatoid arthritis sera boost the Fc receptor- and complement-dependent effector functions of the disease-specific anti-citrullinated protein autoantibodies. J. Immunol. 2015, 194, 3664–3674. [Google Scholar] [CrossRef] [Green Version]
- Laurent, L.; Anquetil, F.; Clavel, C.; Ndongo-Thiam, N.; Offer, G.; Miossec, P.; Pasquali, J.L.; Sebbag, M.; Serre, G. IgM rheumatoid factor amplifies the inflammatory response of macrophages induced by the rheumatoid arthritis-specific immune complexes containing anticitrullinated protein antibodies. Ann. Rheum. Dis. 2015, 74, 1425–1431. [Google Scholar] [CrossRef] [PubMed]
- Sokolove, J.; Johnson, D.S.; Lahey, L.J.; Wagner, C.A.; Cheng, D.; Thiele, G.M.; Michaud, K.; Sayles, H.; Reimold, A.M.; Caplan, L.; et al. Rheumatoid factor as a potentiator of anti-citrullinated protein antibody-mediated inflammation in rheumatoid arthritis. Arthritis Rheumatol. 2014, 66, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Yamakawa, M.; Ouhara, K.; Kajiya, M.; Munenaga, S.; Kittaka, M.; Yamasaki, S.; Takeda, K.; Takeshita, K.; Mizuno, N.; Fujita, T.; et al. Porphyromonas gingivalis infection exacerbates the onset of rheumatoid arthritis in SKG mice. Clin. Exp. Immunol. 2016, 186, 177–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnoux, F.; Mariot, C.; Peen, E.; Lambert, N.C.; Balandraud, N.; Roudier, J.; Auger, I. Peptidyl arginine deiminase immunization induces anticitrullinated protein antibodies in mice with particular MHC types. Proc. Natl. Acad. Sci. USA 2017, 114, E10169–E10177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witalison, E.E.; Thompson, P.R.; Hofseth, L.J. Protein Arginine Deiminases and Associated Citrullination: Physiological Functions and Diseases Associated with Dysregulation. Curr. Drug Targets 2015, 16, 700–710. [Google Scholar] [CrossRef]
- Rogers, G.E.; Harding, H.W.; Llewellyn-Smith, I.J. The origin of citrulline-containing proteins in the hair follicle and the chemical nature of trichohyalin, an intracellular precursor. Biochim. Biophys Acta 1977, 495, 159–175. [Google Scholar] [CrossRef]
- Fujisaki, M.; Sugawara, K. Properties of peptidylarginine deiminase from the epidermis of newborn rats. J. Biochem. 1981, 89, 257–263. [Google Scholar] [CrossRef]
- Cuthbert, G.L.; Daujat, S.; Snowden, A.W.; Erdjument-Bromage, H.; Hagiwara, T.; Yamada, M.; Schneider, R.; Gregory, P.D.; Tempst, P.; Bannister, A.J.; et al. Histone deimination antagonizes arginine methylation. Cell 2004, 118, 545–553. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wysocka, J.; Sayegh, J.; Lee, Y.H.; Perlin, J.R.; Leonelli, L.; Sonbuchner, L.S.; McDonald, C.H.; Cook, R.G.; Dou, Y.; et al. Human PAD4 regulates histone arginine methylation levels via demethylimination. Science 2004, 306, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Tarcsa, E.; Marekov, L.N.; Mei, G.; Melino, G.; Lee, S.C.; Steinert, P.M. Protein unfolding by peptidylarginine deiminase. Substrate specificity and structural relationships of the natural substrates trichohyalin and filaggrin. J. Biol. Chem. 1996, 271, 30709–30716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blachere, N.E.; Parveen, S.; Frank, M.O.; Dill, B.D.; Molina, H.; Orange, D.E. High-Titer Rheumatoid Arthritis Antibodies Preferentially Bind Fibrinogen Citrullinated by Peptidylarginine Deiminase 4. Arthritis Rheumatol. 2017, 69, 986–995. [Google Scholar] [CrossRef]
- Foulquier, C.; Sebbag, M.; Clavel, C.; Chapuy-Regaud, S.; Al Badine, R.; Mechin, M.C.; Vincent, C.; Nachat, R.; Yamada, M.; Takahara, H.; et al. Peptidyl arginine deiminase type 2 (PAD-2) and PAD-4 but not PAD-1, PAD-3, and PAD-6 are expressed in rheumatoid arthritis synovium in close association with tissue inflammation. Arthritis Rheum. 2007, 56, 3541–3553. [Google Scholar] [CrossRef]
- Chang, X.; Yamada, R.; Suzuki, A.; Sawada, T.; Yoshino, S.; Tokuhiro, S.; Yamamoto, K. Localization of peptidylarginine deiminase 4 (PADI4) and citrullinated protein in synovial tissue of rheumatoid arthritis. Rheumatology 2005, 44, 40–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, A.; Yamada, R.; Chang, X.; Tokuhiro, S.; Sawada, T.; Suzuki, M.; Nagasaki, M.; Nakayama-Hamada, M.; Kawaida, R.; Ono, M.; et al. Functional haplotypes of PADI4, encoding citrullinating enzyme peptidylarginine deiminase 4, are associated with rheumatoid arthritis. Nat. Genet. 2003, 34, 395–402. [Google Scholar] [CrossRef]
- Plenge, R.M.; Padyukov, L.; Remmers, E.F.; Purcell, S.; Lee, A.T.; Karlson, E.W.; Wolfe, F.; Kastner, D.L.; Alfredsson, L.; Altshuler, D.; et al. Replication of putative candidate-gene associations with rheumatoid arthritis in >4,000 samples from North America and Sweden: Association of susceptibility with PTPN22, CTLA4, and PADI4. Am. J. Hum. Genet. 2005, 77, 1044–1060. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, Y.; Terao, C. The Impact of Cigarette Smoking on Risk of Rheumatoid Arthritis: A Narrative Review. Cells 2020, 9, 475. [Google Scholar] [CrossRef] [Green Version]
- Kallberg, H.; Ding, B.; Padyukov, L.; Bengtsson, C.; Ronnelid, J.; Klareskog, L.; Alfredsson, L.; Group, E.S. Smoking is a major preventable risk factor for rheumatoid arthritis: Estimations of risks after various exposures to cigarette smoke. Ann. Rheum. Dis. 2011, 70, 508–511. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Ikari, K.; Hashimoto, M.; Ohmura, K.; Tanaka, M.; Ito, H.; Taniguchi, A.; Yamanaka, H.; Mimori, T.; Terao, C. Shared epitope defines distinct associations of cigarette smoking with levels of anticitrullinated protein antibody and rheumatoid factor. Ann. Rheum. Dis. 2019, 78, 1480–1487. [Google Scholar] [CrossRef]
- Gomez-Banuelos, E.; Mukherjee, A.; Darrah, E.; Andrade, F. Rheumatoid Arthritis-Associated Mechanisms of Porphyromonas gingivalis and Aggregatibacter actinomycetemcomitans. J. Clin. Med. 2019, 8, 1309. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhang, D.; Jia, H.; Feng, Q.; Wang, D.; Liang, D.; Wu, X.; Li, J.; Tang, L.; Li, Y.; et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat. Med. 2015, 21, 895–905. [Google Scholar] [CrossRef]
- Chen, J.; Wright, K.; Davis, J.M.; Jeraldo, P.; Marietta, E.V.; Murray, J.; Nelson, H.; Matteson, E.L.; Taneja, V. An expansion of rare lineage intestinal microbes characterizes rheumatoid arthritis. Genome. Med. 2016, 8, 43. [Google Scholar] [CrossRef] [Green Version]
- Maeda, Y.; Kurakawa, T.; Umemoto, E.; Motooka, D.; Ito, Y.; Gotoh, K.; Hirota, K.; Matsushita, M.; Furuta, Y.; Narazaki, M.; et al. Dysbiosis Contributes to Arthritis Development via Activation of Autoreactive T Cells in the Intestine. Arthritis Rheumatol. 2016, 68, 2646–2661. [Google Scholar] [CrossRef] [PubMed]
- Courbon, G.; Rinaudo-Gaujous, M.; Blasco-Baque, V.; Auger, I.; Caire, R.; Mijola, L.; Vico, L.; Paul, S.; Marotte, H. Porphyromonas gingivalis experimentally induces periodontis and an anti-CCP2-associated arthritis in the rat. Ann. Rheum. Dis. 2019, 78, 594–599. [Google Scholar] [CrossRef] [PubMed]
- McGraw, W.T.; Potempa, J.; Farley, D.; Travis, J. Purification, characterization, and sequence analysis of a potential virulence factor from Porphyromonas gingivalis, peptidylarginine deiminase. Infect. Immun. 1999, 67, 3248–3256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Aquino, S.G.; Talbot, J.; Sonego, F.; Turato, W.M.; Grespan, R.; Avila-Campos, M.J.; Cunha, F.Q.; Cirelli, J.A. The aggravation of arthritis by periodontitis is dependent of IL-17 receptor A activation. J. Clin. Periodontol. 2017, 44, 881–891. [Google Scholar] [CrossRef] [PubMed]
- De Aquino, S.G.; Abdollahi-Roodsaz, S.; Koenders, M.I.; van de Loo, F.A.; Pruijn, G.J.; Marijnissen, R.J.; Walgreen, B.; Helsen, M.M.; van den Bersselaar, L.A.; de Molon, R.S.; et al. Periodontal pathogens directly promote autoimmune experimental arthritis by inducing a TLR2- and IL-1-driven Th17 response. J. Immunol. 2014, 192, 4103–4111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spengler, J.; Lugonja, B.; Ytterberg, A.J.; Zubarev, R.A.; Creese, A.J.; Pearson, M.J.; Grant, M.M.; Milward, M.; Lundberg, K.; Buckley, C.D.; et al. Release of Active Peptidyl Arginine Deiminases by Neutrophils Can Explain Production of Extracellular Citrullinated Autoantigens in Rheumatoid Arthritis Synovial Fluid. Arthritis Rheumatol. 2015, 67, 3135–3145. [Google Scholar] [CrossRef] [Green Version]
- Sorice, M.; Iannuccelli, C.; Manganelli, V.; Capozzi, A.; Alessandri, C.; Lococo, E.; Garofalo, T.; Di Franco, M.; Bombardieri, M.; Nerviani, A.; et al. Autophagy generates citrullinated peptides in human synoviocytes: A possible trigger for anti-citrullinated peptide antibodies. Rheumatology 2016, 55, 1374–1385. [Google Scholar] [CrossRef] [Green Version]
- Khandpur, R.; Carmona-Rivera, C.; Vivekanandan-Giri, A.; Gizinski, A.; Yalavarthi, S.; Knight, J.S.; Friday, S.; Li, S.; Patel, R.M.; Subramanian, V.; et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci. Transl. Med. 2013, 5, 178ra140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugawara, E.; Kato, M.; Kudo, Y.; Lee, W.; Hisada, R.; Fujieda, Y.; Oku, K.; Bohgaki, T.; Amengual, O.; Yasuda, S.; et al. Autophagy promotes citrullination of VIM (vimentin) and its interaction with major histocompatibility complex class II in synovial fibroblasts. Autophagy 2019, 16, 946–1955. [Google Scholar] [CrossRef] [PubMed]
- Vomero, M.; Manganelli, V.; Barbati, C.; Colasanti, T.; Capozzi, A.; Finucci, A.; Spinelli, F.R.; Ceccarelli, F.; Perricone, C.; Truglia, S.; et al. Reduction of autophagy and increase in apoptosis correlates with a favorable clinical outcome in patients with rheumatoid arthritis treated with anti-TNF drugs. Arthritis Res. Ther. 2019, 21, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmona-Rivera, C.; Carlucci, P.M.; Moore, E.; Lingampalli, N.; Uchtenhagen, H.; James, E.; Liu, Y.; Bicker, K.L.; Wahamaa, H.; Hoffmann, V.; et al. Synovial fibroblast-neutrophil interactions promote pathogenic adaptive immunity in rheumatoid arthritis. Sci. Immunol. 2017, 2. [Google Scholar] [CrossRef] [Green Version]
- Darrah, E.; Andrade, F. Rheumatoid arthritis and citrullination. Curr. Opin. Rheumatol. 2018, 30, 72–78. [Google Scholar] [CrossRef]
- Kinloch, A.; Lundberg, K.; Wait, R.; Wegner, N.; Lim, N.H.; Zendman, A.J.; Saxne, T.; Malmstrom, V.; Venables, P.J. Synovial fluid is a site of citrullination of autoantigens in inflammatory arthritis. Arthritis Rheum. 2008, 58, 2287–2295. [Google Scholar] [CrossRef]
- Nakayama-Hamada, M.; Suzuki, A.; Kubota, K.; Takazawa, T.; Ohsaka, M.; Kawaida, R.; Ono, M.; Kasuya, A.; Furukawa, H.; Yamada, R.; et al. Comparison of enzymatic properties between hPADI2 and hPADI4. Biochem. Biophys Res. Commun. 2005, 327, 192–200. [Google Scholar] [CrossRef]
- Kinne, R.W.; Brauer, R.; Stuhlmuller, B.; Palombo-Kinne, E.; Burmester, G.R. Macrophages in rheumatoid arthritis. Arthritis Res. 2000, 2, 189–202. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.C.; Lai, N.S.; Yu, H.C.; Huang, H.B.; Hsieh, S.C.; Yu, C.L. Anti-citrullinated protein antibodies bind surface-expressed citrullinated Grp78 on monocyte/macrophages and stimulate tumor necrosis factor alpha production. Arthritis Rheum. 2010, 62, 1213–1223. [Google Scholar] [CrossRef]
- Lu, M.C.; Lai, N.S.; Yin, W.Y.; Yu, H.C.; Huang, H.B.; Tung, C.H.; Huang, K.Y.; Yu, C.L. Anti-citrullinated protein antibodies activated ERK1/2 and JNK mitogen-activated protein kinases via binding to surface-expressed citrullinated GRP78 on mononuclear cells. J. Clin. Immunol. 2013, 33, 558–566. [Google Scholar] [CrossRef]
- Lai, N.S.; Yu, H.C.; Yu, C.L.; Koo, M.; Huang, H.B.; Lu, M.C. Anti-citrullinated protein antibodies suppress let-7a expression in monocytes from patients with rheumatoid arthritis and facilitate the inflammatory responses in rheumatoid arthritis. Immunobiology 2015, 220, 1351–1358. [Google Scholar] [CrossRef] [PubMed]
- Clavel, C.; Nogueira, L.; Laurent, L.; Iobagiu, C.; Vincent, C.; Sebbag, M.; Serre, G. Induction of macrophage secretion of tumor necrosis factor alpha through Fcgamma receptor IIa engagement by rheumatoid arthritis-specific autoantibodies to citrullinated proteins complexed with fibrinogen. Arthritis Rheum. 2008, 58, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Clavel, C.; Ceccato, L.; Anquetil, F.; Serre, G.; Sebbag, M. Among human macrophages polarised to different phenotypes, the M-CSF-oriented cells present the highest pro-inflammatory response to the rheumatoid arthritis-specific immune complexes containing ACPA. Ann. Rheum. Dis. 2016, 75, 2184–2191. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, M.; Baron, B. The role of TNF-alpha in rheumatoid arthritis: A focus on regulatory T cells. J. Clin. Transl. Res. 2016, 2, 84–90. [Google Scholar] [CrossRef]
- Nurcombe, H.L.; Bucknall, R.C.; Edwards, S.W. Neutrophils isolated from the synovial fluid of patients with rheumatoid arthritis: Priming and activation in vivo. Ann. Rheum. Dis. 1991, 50, 147–153. [Google Scholar] [CrossRef] [Green Version]
- Watson, F.; Robinson, J.J.; Phelan, M.; Bucknall, R.C.; Edwards, S.W. Receptor expression in synovial fluid neutrophils from patients with rheumatoid arthritis. Ann. Rheum. Dis. 1993, 52, 354–359. [Google Scholar] [CrossRef] [Green Version]
- Quayle, J.A.; Watson, F.; Bucknall, R.C.; Edwards, S.W. Neutrophils from the synovial fluid of patients with rheumatoid arthritis express the high affinity immunoglobulin G receptor, Fc gamma RI (CD64): Role of immune complexes and cytokines in induction of receptor expression. Immunology 1997, 91, 266–273. [Google Scholar] [CrossRef]
- Robinson, J.J.; Watson, F.; Bucknall, R.C.; Edwards, S.W. Role of Fc gamma receptors in the activation of neutrophils by soluble and insoluble immunoglobulin aggregates isolated from the synovial fluid of patients with rheumatoid arthritis. Ann. Rheum. Dis. 1994, 53, 515–520. [Google Scholar] [CrossRef] [Green Version]
- Kempers, A.C.; Nejadnik, M.R.; Rombouts, Y.; Ioan-Facsinay, A.; van Oosterhout, M.; Jiskoot, W.; Huizinga, T.W.J.; Toes, R.E.M.; Scherer, H.U. Fc gamma receptor binding profile of anti-citrullinated protein antibodies in immune complexes suggests a role for FcgammaRI in the pathogenesis of synovial inflammation. Clin. Exp. Rheumatol. 2018, 36, 284–293. [Google Scholar]
- Wright, H.L.; Moots, R.J.; Edwards, S.W. The multifactorial role of neutrophils in rheumatoid arthritis. Nat. Rev. Rheumatol. 2014, 10, 593–601. [Google Scholar] [CrossRef]
- Chirivi, R.G.S.; van Rosmalen, J.W.G.; van der Linden, M.; Euler, M.; Schmets, G.; Bogatkevich, G.; Kambas, K.; Hahn, J.; Braster, Q.; Soehnlein, O.; et al. Therapeutic ACPA inhibits NET formation: A potential therapy for neutrophil-mediated inflammatory diseases. Cell Mol. Immunol. 2020, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corsiero, E.; Bombardieri, M.; Carlotti, E.; Pratesi, F.; Robinson, W.; Migliorini, P.; Pitzalis, C. Single cell cloning and recombinant monoclonal antibodies generation from RA synovial B cells reveal frequent targeting of citrullinated histones of NETs. Ann. Rheum. Dis. 2016, 75, 1866–1875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloyd, K.A.; Wigerblad, G.; Sahlstrom, P.; Garimella, M.G.; Chemin, K.; Steen, J.; Titcombe, P.J.; Marklein, B.; Zhou, D.; Stalesen, R.; et al. Differential ACPA Binding to Nuclear Antigens Reveals a PAD-Independent Pathway and a Distinct Subset of Acetylation Cross-Reactive Autoantibodies in Rheumatoid Arthritis. Front. Immunol. 2018, 9, 3033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holers, V.M.; Banda, N.K. Complement in the Initiation and Evolution of Rheumatoid Arthritis. Front. Immunol. 2018, 9, 1057. [Google Scholar] [CrossRef]
- Di Muzio, G.; Perricone, C.; Ballanti, E.; Kroegler, B.; Greco, E.; Novelli, L.; Conigliaro, P.; Cipriani, P.; Giacomelli, R.; Perricone, R. Complement system and rheumatoid arthritis: Relationships with autoantibodies, serological, clinical features, and anti-TNF treatment. Int. J. Immunopathol. Pharmacol. 2011, 24, 357–366. [Google Scholar] [CrossRef] [Green Version]
- Sacks, T.; Moldow, C.F.; Craddock, P.R.; Bowers, T.K.; Jacob, H.S. Oxygen radicals mediate endothelial cell damage by complement-stimulated granulocytes. An in vitro model of immune vascular damage. J. Clin. Invest. 1978, 61, 1161–1167. [Google Scholar] [CrossRef] [Green Version]
- Shushakova, N.; Skokowa, J.; Schulman, J.; Baumann, U.; Zwirner, J.; Schmidt, R.E.; Gessner, J.E. C5a anaphylatoxin is a major regulator of activating versus inhibitory FcgammaRs in immune complex-induced lung disease. J. Clin. Invest. 2002, 110, 1823–1830. [Google Scholar] [CrossRef] [Green Version]
- Tsuboi, N.; Ernandez, T.; Li, X.; Nishi, H.; Cullere, X.; Mekala, D.; Hazen, M.; Kohl, J.; Lee, D.M.; Mayadas, T.N. Regulation of human neutrophil Fcgamma receptor IIa by C5a receptor promotes inflammatory arthritis in mice. Arthritis Rheum. 2011, 63, 467–478. [Google Scholar] [CrossRef] [Green Version]
- Aarvak, T.; Natvig, J.B. Cell-cell interactions in synovitis: Antigen presenting cells and T cell interaction in rheumatoid arthritis. Arthritis Res. 2001, 3, 13–17. [Google Scholar] [CrossRef] [Green Version]
- Takemura, S.; Braun, A.; Crowson, C.; Kurtin, P.J.; Cofield, R.H.; O’Fallon, W.M.; Goronzy, J.J.; Weyand, C.M. Lymphoid neogenesis in rheumatoid synovitis. J. Immunol. 2001, 167, 1072–1080. [Google Scholar] [CrossRef] [Green Version]
- Kerkman, P.F.; Kempers, A.C.; van der Voort, E.I.; van Oosterhout, M.; Huizinga, T.W.; Toes, R.E.; Scherer, H.U. Synovial fluid mononuclear cells provide an environment for long-term survival of antibody-secreting cells and promote the spontaneous production of anti-citrullinated protein antibodies. Ann. Rheum. Dis. 2016, 75, 2201–2207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humby, F.; Bombardieri, M.; Manzo, A.; Kelly, S.; Blades, M.C.; Kirkham, B.; Spencer, J.; Pitzalis, C. Ectopic lymphoid structures support ongoing production of class-switched autoantibodies in rheumatoid synovium. PLoS Med. 2009, 6, e1. [Google Scholar] [CrossRef]
- Takemura, S.; Klimiuk, P.A.; Braun, A.; Goronzy, J.J.; Weyand, C.M. T cell activation in rheumatoid synovium is B cell dependent. J. Immunol. 2001, 167, 4710–4718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, E.A.; Rieck, M.; Pieper, J.; Gebe, J.A.; Yue, B.B.; Tatum, M.; Peda, M.; Sandin, C.; Klareskog, L.; Malmstrom, V.; et al. Citrulline-specific Th1 cells are increased in rheumatoid arthritis and their frequency is influenced by disease duration and therapy. Arthritis Rheumatol. 2014, 66, 1712–1722. [Google Scholar] [CrossRef] [PubMed]
- Snir, O.; Rieck, M.; Gebe, J.A.; Yue, B.B.; Rawlings, C.A.; Nepom, G.; Malmstrom, V.; Buckner, J.H. Identification and functional characterization of T cells reactive to citrullinated vimentin in HLA-DRB1*0401-positive humanized mice and rheumatoid arthritis patients. Arthritis Rheum. 2011, 63, 2873–2883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meednu, N.; Zhang, H.; Owen, T.; Sun, W.; Wang, V.; Cistrone, C.; Rangel-Moreno, J.; Xing, L.; Anolik, J.H. Production of RANKL by Memory B Cells: A Link Between B Cells and Bone Erosion in Rheumatoid Arthritis. Arthritis Rheumatol. 2016, 68, 805–816. [Google Scholar] [CrossRef]
- Cope, A.P. T cells in rheumatoid arthritis. Arthritis Res. Ther. 2008, 10, S1. [Google Scholar] [CrossRef] [Green Version]
- Gaffen, S.L. The role of interleukin-17 in the pathogenesis of rheumatoid arthritis. Curr. Rheumatol. Rep. 2009, 11, 365–370. [Google Scholar] [CrossRef]
- Ehrhardt, G.R.; Hijikata, A.; Kitamura, H.; Ohara, O.; Wang, J.Y.; Cooper, M.D. Discriminating gene expression profiles of memory B cell subpopulations. J. Exp. Med. 2008, 205, 1807–1817. [Google Scholar] [CrossRef] [Green Version]
- Pistoia, V. Production of cytokines by human B cells in health and disease. Immunol. Today 1997, 18, 343–350. [Google Scholar] [CrossRef]
- Harre, U.; Georgess, D.; Bang, H.; Bozec, A.; Axmann, R.; Ossipova, E.; Jakobsson, P.J.; Baum, W.; Nimmerjahn, F.; Szarka, E.; et al. Induction of osteoclastogenesis and bone loss by human autoantibodies against citrullinated vimentin. J. Clin. Invest. 2012, 122, 1791–1802. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, A.; Joshua, V.; Haj Hensvold, A.; Jin, T.; Sun, M.; Vivar, N.; Ytterberg, A.J.; Engstrom, M.; Fernandes-Cerqueira, C.; Amara, K.; et al. Identification of a novel chemokine-dependent molecular mechanism underlying rheumatoid arthritis-associated autoantibody-mediated bone loss. Ann. Rheum. Dis. 2016, 75, 721–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wigerblad, G.; Bas, D.B.; Fernades-Cerqueira, C.; Krishnamurthy, A.; Nandakumar, K.S.; Rogoz, K.; Kato, J.; Sandor, K.; Su, J.; Jimenez-Andrade, J.M.; et al. Autoantibodies to citrullinated proteins induce joint pain independent of inflammation via a chemokine-dependent mechanism. Ann. Rheum. Dis. 2016, 75, 730–738. [Google Scholar] [CrossRef] [Green Version]
- Amara, K.; Steen, J.; Murray, F.; Morbach, H.; Fernandez-Rodriguez, B.M.; Joshua, V.; Engstrom, M.; Snir, O.; Israelsson, L.; Catrina, A.I.; et al. Retraction: Monoclonal IgG antibodies generated from joint-derived B cells of RA patients have a strong bias toward citrullinated autoantigen recognition. J. Exp. Med. 2019, 216, 245. [Google Scholar] [CrossRef] [PubMed]
- Correction: Autoantibodies to citrullinated proteins induce joint pain independent of inflammation via a chemokine-dependent mechanism. Ann. Rheum. Dis. 2019, 78, 865. [CrossRef] [PubMed] [Green Version]
- Correction: Identification of a novel chemokine-dependent molecular mechanism underlying rheumatoid arthritis-associated autoantibody-mediated bone loss. Ann. Rheum. Dis. 2019, 78, 866. [CrossRef] [PubMed] [Green Version]
- Toes, R.; Pisetsky, D.S. Pathogenic effector functions of ACPA: Where do we stand? Ann. Rheum. Dis. 2019, 78, 716–721. [Google Scholar] [CrossRef] [Green Version]
- Holmdahl, R. Comment on editorial ’Pathogenic effector functions of ACPA: where do we stand? ’ Ann. Rheum. Dis. 2019. [Google Scholar] [CrossRef]
- Sun, M.; Rethi, B.; Krishnamurthy, A.; Joshua, V.; Circiumaru, A.; Hensvold, A.H.; Ossipova, E.; Gronwall, C.; Liu, Y.; Engstrom, M.; et al. Anticitrullinated protein antibodies facilitate migration of synovial tissue-derived fibroblasts. Ann. Rheum. Dis. 2019, 78, 1621–1631. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, M.B.; Nakamura, M.C. A Comprehensive Review of Immunoreceptor Regulation of Osteoclasts. Clin. Rev. Allergy Immunol. 2016, 51, 48–58. [Google Scholar] [CrossRef] [Green Version]
- Mocsai, A.; Humphrey, M.B.; Van Ziffle, J.A.; Hu, Y.; Burghardt, A.; Spusta, S.C.; Majumdar, S.; Lanier, L.L.; Lowell, C.A.; Nakamura, M.C. The immunomodulatory adapter proteins DAP12 and Fc receptor gamma-chain (FcRgamma) regulate development of functional osteoclasts through the Syk tyrosine kinase. Proc. Natl. Acad. Sci. USA 2004, 101, 6158–6163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grevers, L.C.; de Vries, T.J.; Everts, V.; Verbeek, J.S.; van den Berg, W.B.; van Lent, P.L. Immune complex-induced inhibition of osteoclastogenesis is mediated via activating but not inhibitory Fcgamma receptors on myeloid precursor cells. Ann. Rheum. Dis. 2013, 72, 278–285. [Google Scholar] [CrossRef]
- Seeling, M.; Hillenhoff, U.; David, J.P.; Schett, G.; Tuckermann, J.; Lux, A.; Nimmerjahn, F. Inflammatory monocytes and Fcgamma receptor IV on osteoclasts are critical for bone destruction during inflammatory arthritis in mice. Proc. Natl. Acad. Sci. USA 2013, 110, 10729–10734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harre, U.; Lang, S.C.; Pfeifle, R.; Rombouts, Y.; Fruhbeisser, S.; Amara, K.; Bang, H.; Lux, A.; Koeleman, C.A.; Baum, W.; et al. Glycosylation of immunoglobulin G determines osteoclast differentiation and bone loss. Nat. Commun. 2015, 6, 6651. [Google Scholar] [CrossRef]
- Hecht, C.; Englbrecht, M.; Rech, J.; Schmidt, S.; Araujo, E.; Engelke, K.; Finzel, S.; Schett, G. Additive effect of anti-citrullinated protein antibodies and rheumatoid factor on bone erosions in patients with RA. Ann. Rheum. Dis. 2015, 74, 2151–2156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bugatti, S.; Bogliolo, L.; Vitolo, B.; Manzo, A.; Montecucco, C.; Caporali, R. Anti-citrullinated protein antibodies and high levels of rheumatoid factor are associated with systemic bone loss in patients with early untreated rheumatoid arthritis. Arthritis Res. Ther. 2016, 18, 226. [Google Scholar] [CrossRef] [Green Version]
- Yoshitomi, H. Regulation of Immune Responses and Chronic Inflammation by Fibroblast-Like Synoviocytes. Front. Immunol. 2019, 10, 1395. [Google Scholar] [CrossRef] [PubMed]
- Corsiero, E.; Jagemann, L.; Perretti, M.; Pitzalis, C.; Bombardieri, M. Characterization of a Synovial B Cell-Derived Recombinant Monoclonal Antibody Targeting Stromal Calreticulin in the Rheumatoid Joints. J. Immunol. 2018, 201, 1373–1381. [Google Scholar] [CrossRef] [PubMed]
- Buckwalter, J.A.; Mankin, H.J. Articular cartilage: Tissue design and chondrocyte-matrix interactions. Instr. Course. Lect. 1998, 47, 477–486. [Google Scholar] [PubMed]
- Beard, H.K.; Ryvar, R.; Skingle, J.; Greenbury, C.L. Anti-collagen antibodies in sera from rheumatoid arthritis patients. J. Clin. Pathol. 1980, 33, 1077–1081. [Google Scholar] [CrossRef] [Green Version]
- Cooke, T.D.; Hurd, E.R.; Jasin, H.E.; Bienenstock, J.; Ziff, M. Identification of immunoglobulins and complement in rheumatoid articular collagenous tissues. Arthritis Rheum. 1975, 18, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.S.; Landewe, R.B.M.; Bijlsma, J.W.J.; Burmester, G.R.; Dougados, M.; Kerschbaumer, A.; McInnes, I.B.; Sepriano, A.; van Vollenhoven, R.F.; de Wit, M.; et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2019 update. Ann. Rheum. Dis. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenzetti, R.; Janowska, I.; Smulski, C.R.; Frede, N.; Henneberger, N.; Walter, L.; Schleyer, M.T.; Huppe, J.M.; Staniek, J.; Salzer, U.; et al. Abatacept modulates CD80 and CD86 expression and memory formation in human B-cells. J. Autoimmun. 2019, 101, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Bohler, C.; Radner, H.; Smolen, J.S.; Aletaha, D. Serological changes in the course of traditional and biological disease modifying therapy of rheumatoid arthritis. Ann. Rheum. Dis. 2013, 72, 241–244. [Google Scholar] [CrossRef] [Green Version]
- Cambridge, G.; Perry, H.C.; Nogueira, L.; Serre, G.; Parsons, H.M.; De La Torre, I.; Dickson, M.C.; Leandro, M.J.; Edwards, J.C. The effect of B-cell depletion therapy on serological evidence of B-cell and plasmablast activation in patients with rheumatoid arthritis over multiple cycles of rituximab treatment. J. Autoimmun. 2014, 50, 67–76. [Google Scholar] [CrossRef]
- Bugatti, S.; Manzo, A.; Montecucco, C.; Caporali, R. The Clinical Value of Autoantibodies in Rheumatoid Arthritis. Front. Med. (Lausanne) 2018, 5, 339. [Google Scholar] [CrossRef]
- Benham, H.; Nel, H.J.; Law, S.C.; Mehdi, A.M.; Street, S.; Ramnoruth, N.; Pahau, H.; Lee, B.T.; Ng, J.; Brunck, M.E.; et al. Citrullinated peptide dendritic cell immunotherapy in HLA risk genotype-positive rheumatoid arthritis patients. Sci. Transl. Med. 2015, 7, 290ra287. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Xia, Y. Anti-double Stranded DNA Antibodies: Origin, Pathogenicity, and Targeted Therapies. Front. Immunol. 2019, 10, 1667. [Google Scholar] [CrossRef] [Green Version]
- Gesheva, V.; Kerekov, N.; Nikolova, K.; Mihaylova, N.; Todorov, T.; Nikolova, M.; Tchorbanov, A. Suppression of dsDNA-specific B lymphocytes reduces disease symptoms in SCID model of mouse lupus. Autoimmunity 2014, 47, 162–172. [Google Scholar] [CrossRef]
- Sthoeger, Z.; Sharabi, A.; Mozes, E. Novel approaches to the development of targeted therapeutic agents for systemic lupus erythematosus. J. Autoimmun. 2014, 54, 60–71. [Google Scholar] [CrossRef]
- Fernandes-Cerqueira, C.; Ossipova, E.; Gunasekera, S.; Hansson, M.; Mathsson, L.; Catrina, A.I.; Sommarin, Y.; Klareskog, L.; Lundberg, K.; Ronnelid, J.; et al. Targeting of anti-citrullinated protein/peptide antibodies in rheumatoid arthritis using peptides mimicking endogenously citrullinated fibrinogen antigens. Arthritis Res. Ther. 2015, 17, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunasekera, S.; Fernandes-Cerqueira, C.; Wennmalm, S.; Wahamaa, H.; Sommarin, Y.; Catrina, A.I.; Jakobsson, P.J.; Goransson, U. Stabilized Cyclic Peptides as Scavengers of Autoantibodies: Neutralization of Anticitrullinated Protein/Peptide Antibodies in Rheumatoid Arthritis. ACS Chem. Biol. 2018, 13, 1525–1535. [Google Scholar] [CrossRef] [PubMed]
- Willis, V.C.; Gizinski, A.M.; Banda, N.K.; Causey, C.P.; Knuckley, B.; Cordova, K.N.; Luo, Y.; Levitt, B.; Glogowska, M.; Chandra, P.; et al. N-alpha-benzoyl-N5-(2-chloro-1-iminoethyl)-L-ornithine amide, a protein arginine deiminase inhibitor, reduces the severity of murine collagen-induced arthritis. J. Immunol. 2011, 186, 4396–4404. [Google Scholar] [CrossRef] [PubMed]
- Aliko, A.; Kaminska, M.; Falkowski, K.; Bielecka, E.; Benedyk-Machaczka, M.; Malicki, S.; Koziel, J.; Wong, A.; Bryzek, D.; Kantyka, T.; et al. Discovery of Novel Potential Reversible Peptidyl Arginine Deiminase Inhibitor. Int. J. Mol. Sci. USA 2019, 20, 2174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reily, C.; Stewart, T.J.; Renfrow, M.B.; Novak, J. Glycosylation in health and disease. Nat. Rev. Nephrol. 2019, 15, 346–366. [Google Scholar] [CrossRef] [PubMed]
- Crivianu-Gaita, V.; Thompson, M. Aptamers, antibody scFv, and antibody Fab’ fragments: An overview and comparison of three of the most versatile biosensor biorecognition elements. Biosens. Bioelectron. 2016, 85, 32–45. [Google Scholar] [CrossRef]
- Hair, P.S.; Enos, A.I.; Krishna, N.K.; Cunnion, K.M. Inhibition of Immune Complex Complement Activation and Neutrophil Extracellular Trap Formation by Peptide Inhibitor of Complement C1. Front. Immunol. 2018, 9, 558. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| ACPA | RF | |
|---|---|---|
| Isotypes | mainly IgG and IgA | IgM > IgG > IgA |
| Clinical association | specific for RA | common in various autoimmune diseases |
| N-glycosylation | extensive | limited |
| Germinal center reactions | repeated | limited |
| Somatic hypermutations | extensive | limited |
| Class switching | extensive | limited |
| B cell activation | T cell dependent | T cell dependent and/or T cell independent |
| Producing plasma cells | long lived plasma cells | short lived plasma cells and/or plasmablasts |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, C.-Y.; Yang, H.-Y.; Lai, J.-H. Anti-Citrullinated Protein Antibodies in Patients with Rheumatoid Arthritis: Biological Effects and Mechanisms of Immunopathogenesis. Int. J. Mol. Sci. 2020, 21, 4015. https://doi.org/10.3390/ijms21114015
Wu C-Y, Yang H-Y, Lai J-H. Anti-Citrullinated Protein Antibodies in Patients with Rheumatoid Arthritis: Biological Effects and Mechanisms of Immunopathogenesis. International Journal of Molecular Sciences. 2020; 21(11):4015. https://doi.org/10.3390/ijms21114015
Chicago/Turabian StyleWu, Chao-Yi, Huang-Yu Yang, and Jenn-Haung Lai. 2020. "Anti-Citrullinated Protein Antibodies in Patients with Rheumatoid Arthritis: Biological Effects and Mechanisms of Immunopathogenesis" International Journal of Molecular Sciences 21, no. 11: 4015. https://doi.org/10.3390/ijms21114015