Blockade of the Adenosine A3 Receptor Attenuates Caspase 1 Activation in Renal Tubule Epithelial Cells and Decreases Interleukins IL-1β and IL-18 in Diabetic Rats

Abstract
:1. Introduction
2. Results
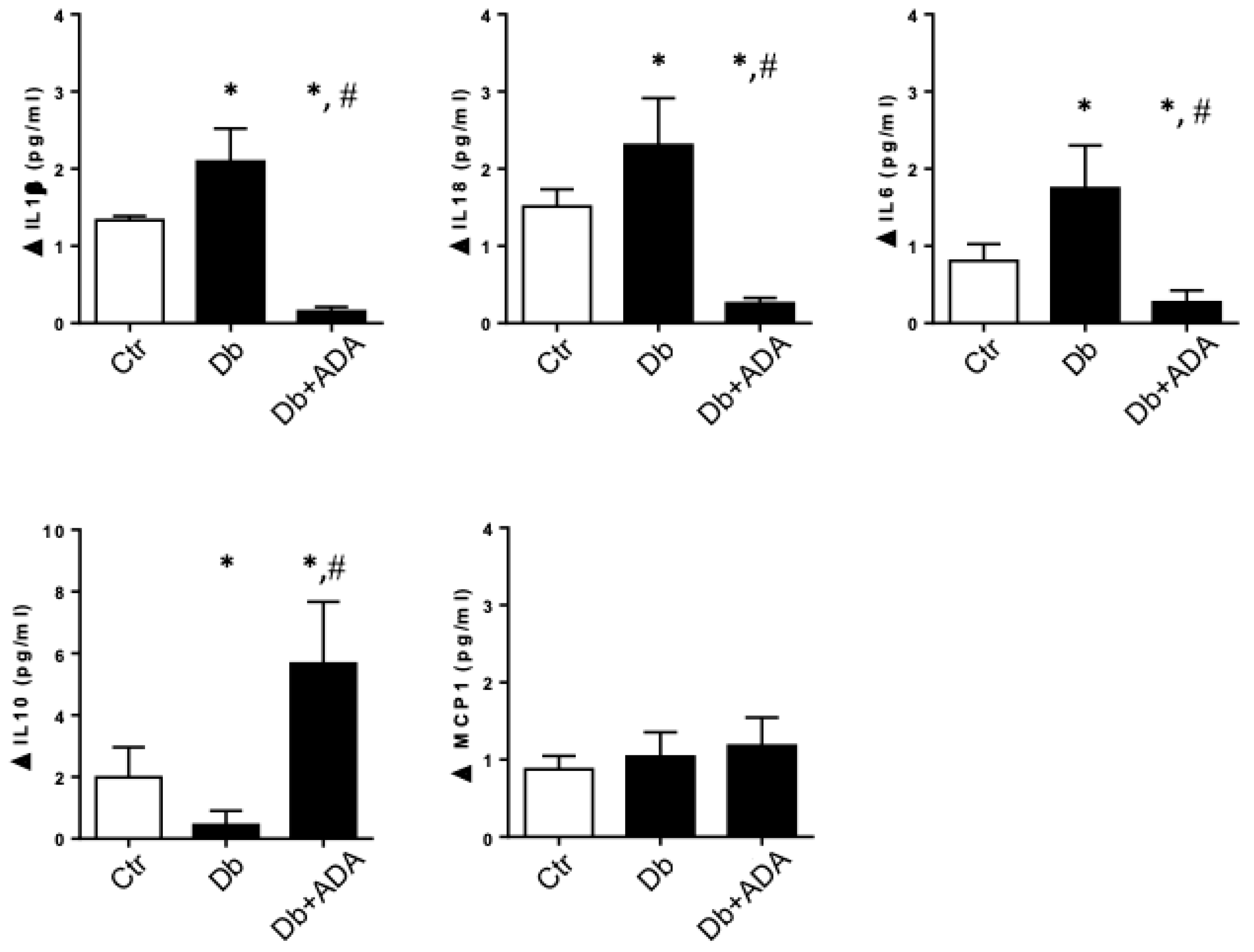
2.1. Adenosine Deaminase Administered in Vivo Affects the Urinary Profile of Cytokines in Diabetic Rats
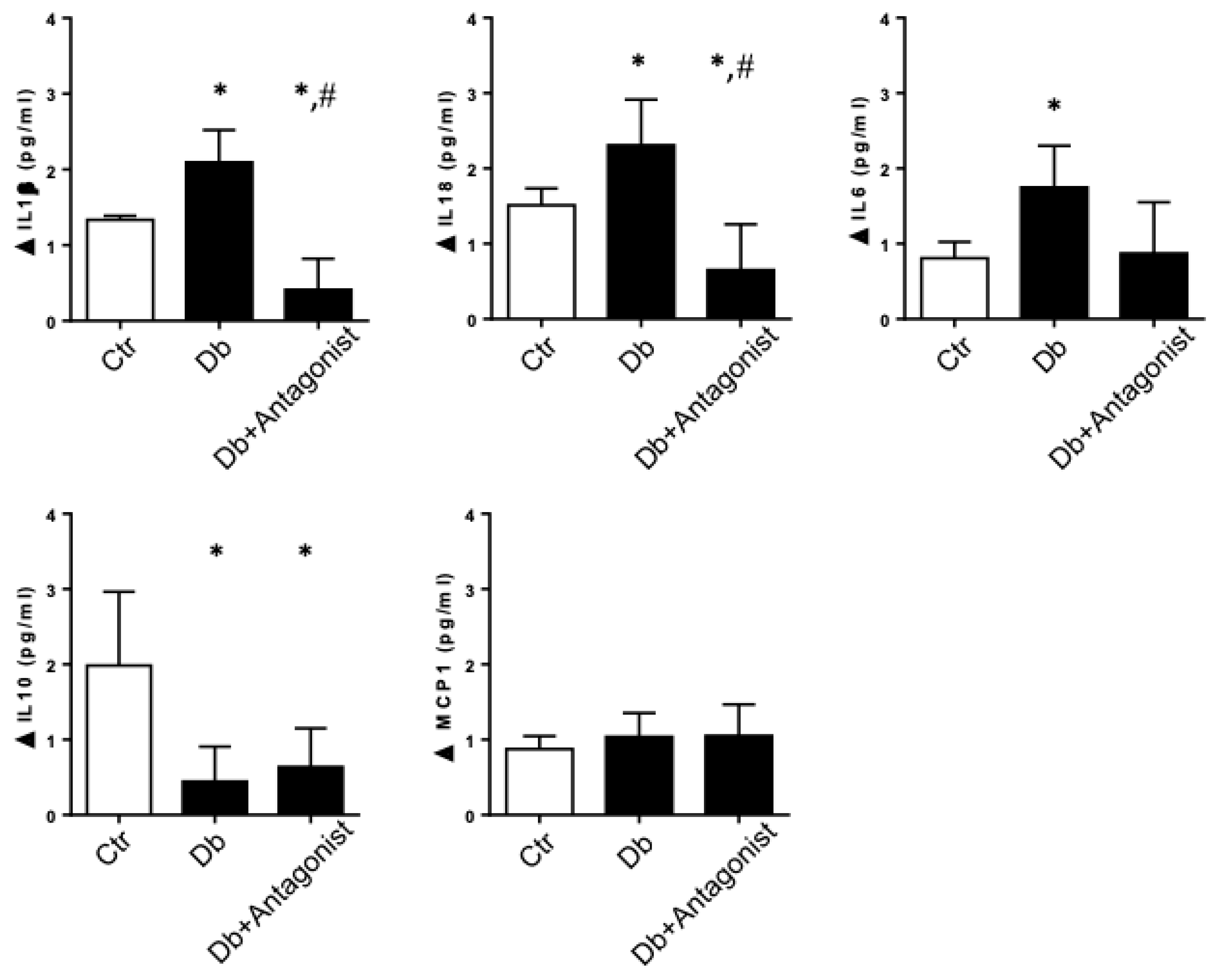
2.2. The ADORA3 Antagonism Decreases the Urine Content of IL-1β and IL18 in Diabetic Rats
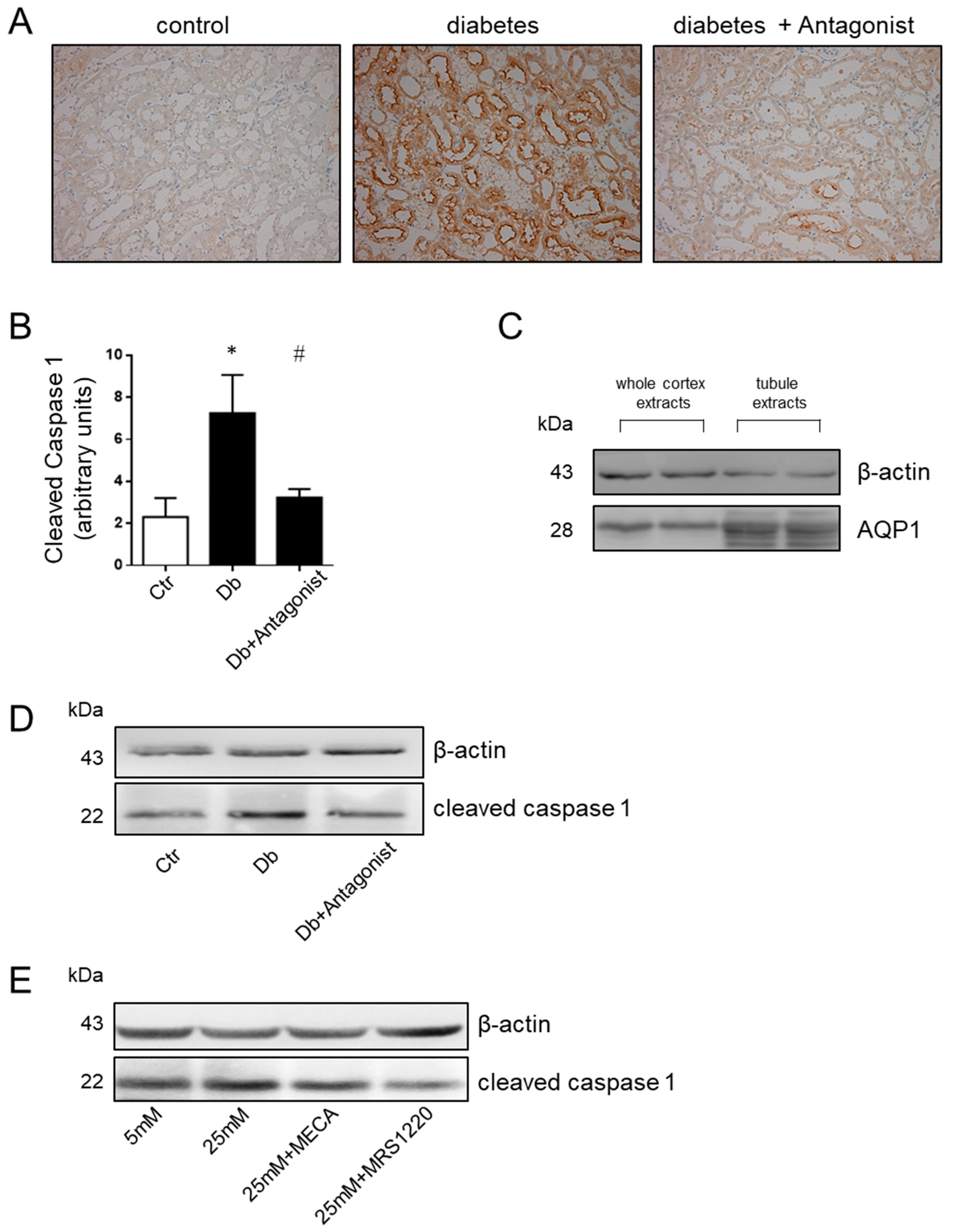
2.3. The ADORA3 Antagonism Affects Selectively Caspase 1 Activation
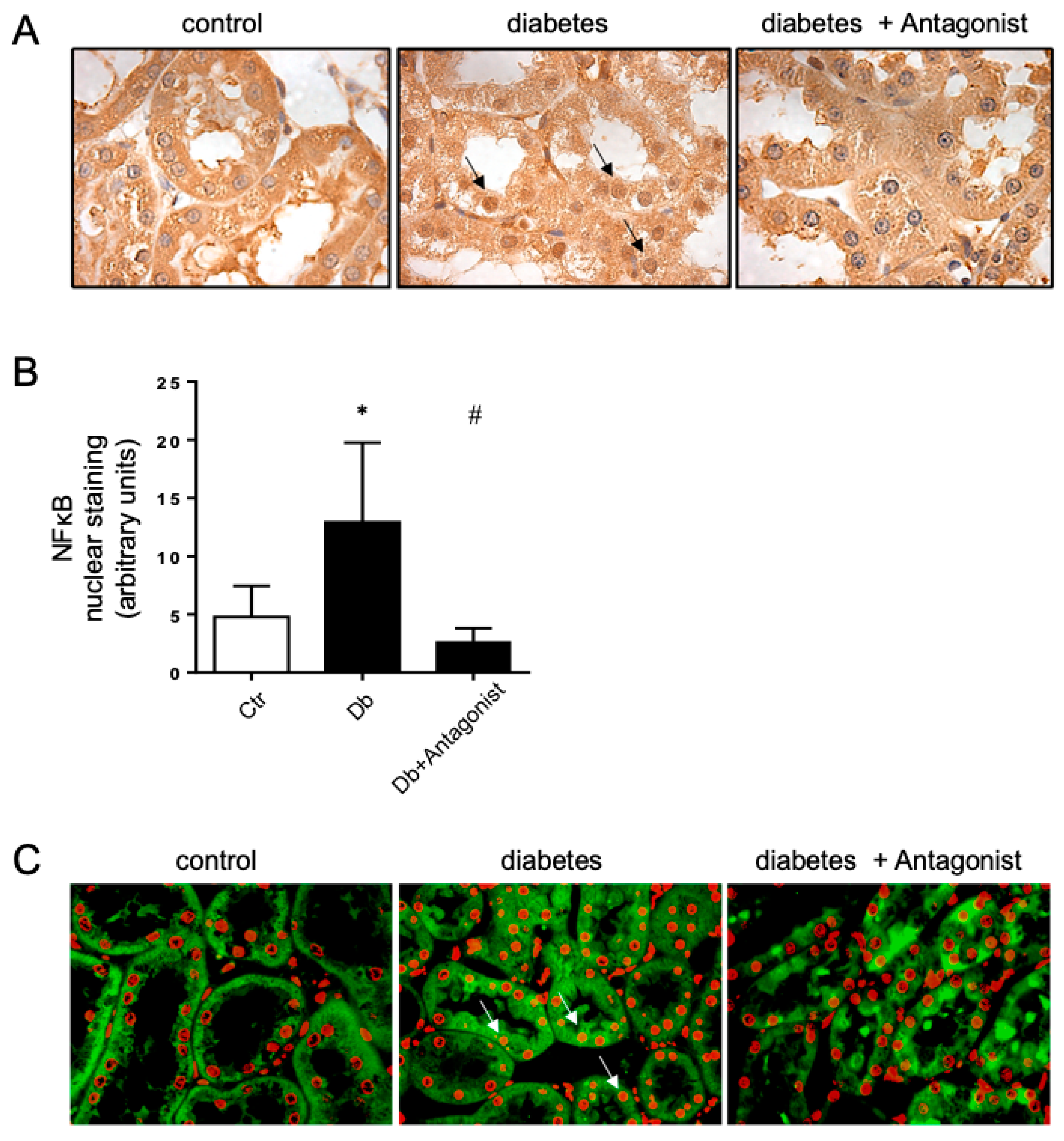
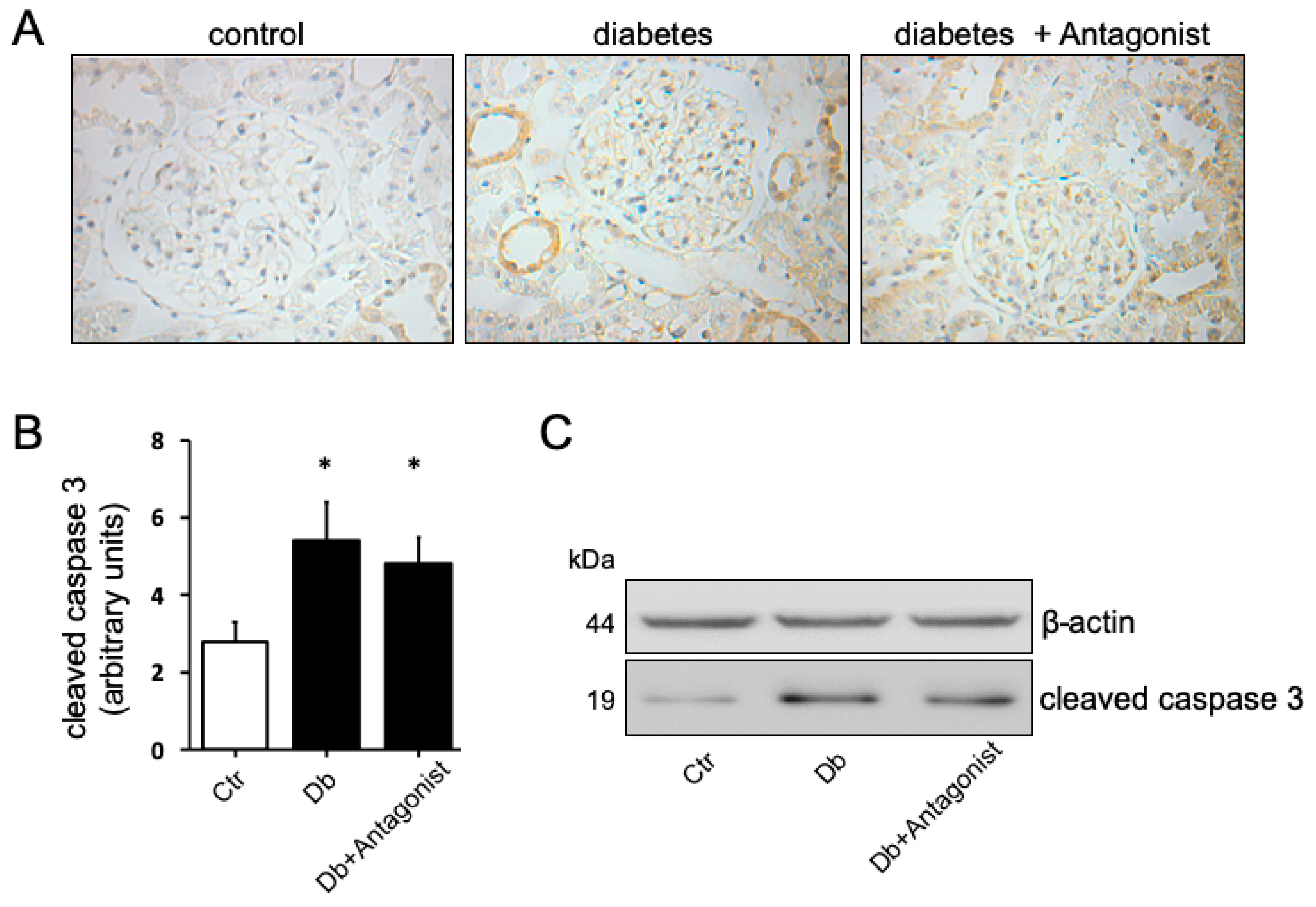
2.4. The Antagonist of the ADORA3 Attenuates Renal Injury in Diabetic Rats
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Animals Models
4.3. Animal Treatments
4.4. Ethics Statement
4.5. Quantification of Cytokines in Urine
4.6. Proteinuria
4.7. Histological Analysis
4.8. The isolation of Proximal Tubules from Rats
4.9. HK2 Cells Treatments
4.10. Western Blots
4.11. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Yach, D.; Stuckler, D.; Brownell, K.D. Epidemiologic and economic consequences of the global epidemics of obesity and diabetes. Nat. Med. 2006, 12, 62–66. [Google Scholar] [CrossRef] [PubMed]
- NCD Risk Factor Collaboration (NCD-RisC). Worldwide trends in diabetes since 1980: A pooled analysis of 751 population-based studies with 4.4 million participants. Lancet 2016, 387, 1513–1530. [Google Scholar] [CrossRef]
- Gross, M.; Dikow, R.; Ritz, E. Diabetic nephropathy: Recent insights into the pathophysiology and the progression of diabetic nephropathy. Kidney Int. Suppl. 2005, 94, S50–S53. [Google Scholar] [CrossRef] [PubMed]
- National Kidney, F. KDOQI Clinical Practice Guideline for Diabetes and CKD: 2012 Update. Am. J. Kidney Dis. 2012, 60, 850–886. [Google Scholar]
- De Boer, I.H.; Rue, T.C.; Hall, Y.N.; Heagerty, P.J.; Weiss, N.S.; Himmelfarb, J. Temporal trends in the prevalence of diabetic kidney disease in the United States. JAMA 2011, 305, 2532–2539. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Fernandez, B.; Ortiz, A.; Gomez-Guerrero, C.; Egido, J. Therapeutic approaches to diabetic nephropathy—beyond the RAS. Nat. Rev. Nephrol. 2014, 10, 325–346. [Google Scholar] [CrossRef] [PubMed]
- Barceló, A.; Aedo, C.; Rajpathak, S.; Robles, S. The cost of diabetes in Latin America and the Caribbean. Bull. World Health Organ. 2003, 81, 19–27. [Google Scholar]
- Mogensen, C.E. Early glomerular hyperfiltration in insulin-dependent diabetics and late nephropathy. Scand. J. Clin. Lab. Invest. 1986, 46, 201–206. [Google Scholar] [CrossRef]
- Gnudi, L. Cellular and molecular mechanisms of diabetic glomerulopathy. Nephrol. Dial. Transpl. 2012, 27, 2642–2649. [Google Scholar] [CrossRef] [Green Version]
- Jermendy, G.; Ruggenenti, P. Preventing microalbuminuria in patients with type 2 diabetes. Diabetes Metab. Res. Rev. 2007, 23, 100–110. [Google Scholar] [CrossRef]
- Nath, K.A. The tubulointerstitium in progressive renal disease. Kidney Int. 1998, 54, 992–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grande, M.T.; Sánchez-Laorden, B.; López-Blau, C.; De Frutos, C.A.; Boutet, A.; Arévalo, M.; Rowe, R.G.; Weiss, S.J.; López-Novoa, J.M.; Nieto, M.A. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 2015, 21, 989–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boor, P.; Ostendorf, T.; Floege, J. Renal fibrosis: Novel insights into mechanisms and therapeutic targets. Nat. Rev. Nephrol. 2010, 6, 643–656. [Google Scholar] [CrossRef] [PubMed]
- Hills, C.E.; Squires, P.E. TGF-beta1-induced epithelial-to-mesenchymal transition; and therapeutic intervention in diabetic nephropathy. Am. J. Nephrol. 2010, 31, 68–74. [Google Scholar] [CrossRef]
- Vega, G.; Alarcon, S.; San Martin, R. The cellular and signalling alterations conducted by TGF-beta contributing to renal fibrosis. Cytokine 2016, 88, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Campanholle, G.; Ligresti, G.; Gharib, S.A.; Duffield, J.S. Cellular mechanisms of tissue fibrosis. 3. Novel mechanisms of kidney fibrosis. Am. J. Physiol. Cell Physiol. 2013, 304, C591–C603. [Google Scholar] [CrossRef]
- Hirschberg, R. Wound healing in the kidney: Complex interactions in renal interstitial fibrogenesis. J. Am. Soc. Nephrol. 2005, 16, 9–11. [Google Scholar] [CrossRef]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. Inflammatory processes in renal fibrosis. Nat. Rev. Nephrol. 2014, 10, 493–503. [Google Scholar] [CrossRef]
- Duffield, J.S.; Lupher, M.; Thannickal, V.J.; Wynn, T.A. Host responses in tissue repair and fibrosis. Annu. Rev. Pathol. 2013, 8, 241–276. [Google Scholar] [CrossRef]
- Bondi, C.D.; Manickam, N.; Lee, D.Y.; Block, K.; Gorin, Y.; Abboud, H.E.; Barnes, J.L. NAD(P)H oxidase mediates TGF-beta1-induced activation of kidney myofibroblasts. J. Am. Soc. Nephrol. 2010, 21, 93–102. [Google Scholar] [CrossRef]
- Chen, Y.T.; Chang, F.C.; Wu, C.F.; Chou, Y.H.; Hsu, H.L.; Chiang, W.C.; Shen, J.; Chen, Y.M.; Wu, K.D.; Tsai, T.J.; et al. Platelet-derived growth factor receptor signaling activates pericyte-myofibroblast transition in obstructive and post-ischemic kidney fibrosis. Kidney Int. 2011, 80, 1170–1181. [Google Scholar] [CrossRef] [PubMed]
- Song, J. EMT or apoptosis: A decision for TGF-beta. Cell Res. 2007, 17, 289–290. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; Taduri, G.; O’Connell, J.; Teng, Y.; Cooke, V.G.; Woda, C.; Sugimoto, H.; Kalluri, R. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 2013, 8, 1047–1053. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.L.; Kisseleva, T.; Brenner, D.A.; Duffield, J.S. Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am. J. Pathol. 2008, 73, 1617–1627. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.F.; Liang, Q.L.; Liang, X.P.; Wang, Y.M.; Hu, P.; Li, P.; Luo, G.A. Ultraviolet and tandem mass spectrometry for simultaneous quantification of 21 pivotal metabolites in plasma from patients with diabetic nephropathy. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2009, 877, 1930–1936. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.F.; Hu, P.; Liang, Q.L.; Zou, T.T.; Wang, Y.M.; Luo, G.A. Correlations of creatine and six related pyrimidine metabolites and diabetic nephropathy in Chinese type 2 diabetic patients. Clin. Biochem. 2010, 43, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Oyarzún, C.; Garrido, W.; Alarcón, S.; Yáñez, A.; Sobrevia, L.; Quezada, C.; San Martín, R. Adenosine contribution to normal renal physiology and chronic kidney disease. Mol. Asp. Med. 2017, 55, 75–89. [Google Scholar] [CrossRef] [Green Version]
- Fredholm, B.B.; Ijzerman, A.P.; Jacobson, K.A.; Linden, J.; Müller, C.E. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors an update. Pharm. Rev. 2011, 63, 1–34. [Google Scholar] [CrossRef]
- Kretschmar, C.; Oyarzún, C.; Villablanca, C.; Jaramillo, C.; Alarcón, S.; Perez, G.; Díaz-Encarnación, M.M.; Pastor-Anglada, M.; Garrido, W.; Quezada, C.; et al. Reduced Adenosine Uptake and Its Contribution to Signaling that Mediates Profibrotic Activation in Renal Tubular Epithelial Cells: Implication in Diabetic Nephropathy. PLoS ONE 2016, 11, e0147430. [Google Scholar] [CrossRef]
- Min, H.S.; Cha, J.J.; Kim, K.; Kim, J.E.; Ghee, J.Y.; Kim, H.; Lee, J.E.; Han, J.Y.; Jeong, L.S.; Cha, D.R.; et al. Renoprotective Effects of a Highly Selective A3 Adenosine Receptor Antagonist in a Mouse Model of Adriamycin-induced Nephropathy. J. Korean Med. Sci 2016, 31, 1403–1412. [Google Scholar] [CrossRef] [Green Version]
- Oyarzún, C.; Salinas, C.; Gómez, D.; Jaramillo, K.; Pérez, G.; Alarcón, S.; Podestá, L.; Flores, C.; Quezada, C.; San Martín, R. Increased levels of adenosine and ecto 5′-nucleotidase (CD73) activity precede renal alterations in experimental diabetic rats. Biochem. Biophys. Res. Commun. 2015, 468, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Rieder, F.; Kessler, S.P.; West, G.A.; Bhilocha, S.; De la Motte, C.; Sadler, T.M.; Gopalan, B.; Stylianou, E.; Fiocchi, C. Inflammation-induced endothelial-to-mesenchymal transition: A novel mechanism of intestinal fibrosis. Am. J. Pathol. 2011, 179, 2660–2673. [Google Scholar] [CrossRef] [PubMed]
- Navarro, J.F.; Mora-Fernandez, C. The role of TNF-alpha in diabetic nephropathy: Pathogenic and therapeutic implications. Cytokine Growth Factor Rev. 2006, 17, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Zhang, W.; Wen, J.; Zhang, Y.; Kellems, R.E.; Xia, Y. A2B adenosine receptor-mediated induction of IL-6 promotes CKD. J. Am. Soc. Nephrol. 2011, 22, 890–901. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Wu, N.; Zhao, D. Function of NLRP3 in the Pathogenesis and Development of Diabetic Nephropathy. Med. Sci. Monit. 2017, 23, 3878–3884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutton, H.L.; Ooi, J.D.; Holdsworth, S.R.; Kitching, A.R. The NLRP3 inflammasome in kidney disease and autoimmunity. Nephrology (Carlton) 2016, 21, 736–744. [Google Scholar] [CrossRef] [PubMed]
- Shahzad, K.; Bock, F.; Dong, W.; Wang, H.; Kopf, S.; Kohli, S.; Al-Dabet, M.M.; Ranjan, S.; Wolter, J.; Wacker, C.; et al. Nlrp3-inflammasome activation in non-myeloid-derived cells aggravates diabetic nephropathy. Kidney Int. 2015, 87, 74–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobson, K.A.; Merighi, S.; Varani, K.; Borea, P.A.; Baraldi, S.; Aghazadeh-Tabrizi, M.; Romagnoli, R.; Baraldi, P.G.; Ciancetta, A.; Tosh, D.K.; et al. A3 Adenosine Receptors as Modulators of Inflammation: From Medicinal Chemistry to Therapy. Med. Res. Rev. 2018, 38, 1031–1072. [Google Scholar] [CrossRef] [PubMed]
- Alarcón, S.; Garrido, W.; Cappelli, C.; Suárez, R.; Oyarzún, C.; Quezada, C.; San Martín, R. Deficient Insulin-mediated Upregulation of the Equilibrative Nucleoside Transporter 2 Contributes to Chronically Increased Adenosine in Diabetic Glomerulopathy. Sci. Rep. 2017, 7, 9439–9445. [Google Scholar] [CrossRef] [PubMed]
- Dorotea, D.; Cho, A.; Lee, G.; Kwon, G.; Lee, J.; Sahu, P.K.; Jeong, L.S.; Cha, D.R.; Ha, H. Orally active, species-independent novel A3 adenosine receptor antagonist protects against kidney injury in db/db mice. Exp. Mol. Med. 2018, 50, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Miyauchi, K.; Takiyama, Y.; Honjyo, J.; Tateno, M.; Haneda, M. Upregulated IL-18 expression in type 2 diabetic subjects with nephropathy: TGF-beta1 enhanced IL-18 expression in human renal proximal tubular epithelial cells. Diabetes Res. Clin. Pr. 2009, 83, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, Y.; Bajaj, M.; Yang, H.C.; Ye, Y. Combined SGLT2 and DPP4 Inhibition Reduces the Activation of the Nlrp3/ASC Inflammasome and Attenuates the Development of Diabetic Nephropathy in Mice with Type 2 Diabetes. Cardiovasc. Drugs Ther. 2018, 32, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Wei, G.; Xu, J.; Ma, X.; Wang, Q. Naringin ameliorates the high glucose-induced rat mesangial cell inflammatory reaction by modulating the NLRP3 Inflammasome. BMC Complement. Altern. Med. 2018, 18, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Shahzad, K.; Bock, F.; Al-Dabet, M.M.; Gadi, I.; Nazir, S.; Wang, H.; Kohli, S.; Ranjan, S.; Mertens, P.R.; Nawroth, P.P.; et al. Stabilization of endogenous Nrf2 by minocycline protects against Nlrp3-inflammasome induced diabetic nephropathy. Sci. Rep. 2016, 6, 34228–34235. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, X.; Ghani, A.; Malik, A.; Wilder, T.; Colegio, O.R.; Flavell, R.A.; Cronstein, B.N.; Mehal, W.Z. Adenosine is required for sustained inflammasome activation via the A₂A receptor and the HIF-1α pathway. Nat. Commun. 2013, 4, 2909–2915. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Shikata, K.; Hiramatsu, M.; Nakatou, T.; Kitamura, T.; Wada, J.; Itoshima, T.; Makino, H. Serum interleukin-18 levels are associated with nephropathy and atherosclerosis in Japanese patients with type 2 diabetes. Diabetes Care 2005, 28, 2890–2895. [Google Scholar] [CrossRef]
- Arellano-Buendía, A.S.; Tostado-González, M.; García-Arroyo, F.E.; Cristíbal-García, M.; Loredo-Mendoza, M.L.; Tapia, E.; Sánchez-Lozada, L.G.; Osorio-Alonso, H. Anti- Inflammatory Therapy Modulates Nrf2-Keap1 in Kidney from Rats with Diabetes. Oxid. Med. Cell. Longev. 2016, 2016, 4693801–4693812. [Google Scholar] [CrossRef]
- Jones, L.K.; O’Sullivan, K.M.; Semple, T.; Kuligowski, M.P.; Fukami, K.; Ma, F.Y.; Nikolic-Paterson, D.J.; Holdsworth, S.R.; Kitching, A.R. IL-1RI deficiency ameliorates early experimental renal interstitial fibrosis. Nephrol. Dial. Transpl. 2009, 24, 3024–3032. [Google Scholar] [CrossRef] [Green Version]
- Bani-Hani, A.H.; Leslie, J.A.; Asanuma, H.; Dinarello, C.A.; Campbell, M.T.; Meldrum, D.R.; Zhang, H.; Hile, K.; Meldrum, K.K. IL-18 neutralization ameliorates obstruction-induced epithelial-mesenchymal transition and renal fibrosis. Kidney Int. 2009, 76, 500–511. [Google Scholar] [CrossRef]
- Wu, H.; Craft, M.L.; Wang, P.; Wyburn, K.R.; Chen, G.; Ma, J.; Hambly, B.; Chadban, S.J. IL-18 contributes to renal damage after ischemia–reperfusion. J. Am. Soc. Nephrol. 2008, 19, 2331–2341. [Google Scholar] [CrossRef]
- Ji, X.; Li, C.; Ou, Y.; Li, N.; Yuan, K.; Yang, G.; Chen, X.; Yang, Z.; Liu, B.; Cheung, W.W.; et al. Andrographolide ameliorates diabetic nephropathy by attenuating hyperglycemia-mediated renal oxidative stress and inflammation via Akt/NF-kappaB pathway. Mol. Cell Endocrinol. 2016, 437, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Kuhad, A.; Chopra, K. Attenuation of diabetic nephropathy by tocotrienol: involvement of NFkB signaling pathway. Life Sci. 2009, 84, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Schmid, H.; Boucherot, A.; Yasuda, Y.; Henger, A.; Brunner, B.; Eichinger, F.; Nitsche, A.; Kiss, E.; Bleich, M.; Gröne, H.J.; et al. European Renal cDNA Bank (ERCB) Consortium. Modular activation of nuclear factor-kappaB transcriptional programs in human diabetic nephropathy. Diabetes 2006, 55, 2993–3003. [Google Scholar] [CrossRef] [PubMed]
- Pawelczyk, T.; Grden, M.; Rzepko, R.; Sakowicz, M.; Szutowicz, A. Region-specific alterations of adenosine receptors expression level in kidney of diabetic rat. Am. J. Pathol. 2005, 167, 315–325. [Google Scholar] [CrossRef]
- Mundell, S.; Kelly, E. Adenosine receptor desensitization and trafficking. Biochim. Biophys. Acta 2011, 1808, 1319–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duann, P.; Ho, T.Y.; Desai, B.D.; Kapoian, T.; Cowen, D.S.; Lianos, E.A. Mesangial cell apoptosis induced by stimulation of the adenosine A3 receptor: Signaling and apoptotic events. J. Investig. Med. 2005, 1, 37–43. [Google Scholar] [CrossRef]
- Sun, W.; Khoo, H.E.; Tan, C.H. Adenosine induced apoptosis in BHK cells via P1 receptors and equilibrative nucleoside transporters. J. Biochem. Mol. Biol. 2005, 38, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Hwang, I.; Lee, J.H.; Lee, H.W.; Jeong, L.S.; Ha, H. The selective A3AR antagonist LJ-1888 ameliorates UUO-induced tubulointerstitial fibrosis. Am. J. Pathol. 2013, 183, 1488–1497. [Google Scholar] [CrossRef] [PubMed]
- Vinay, P.; Gougoux, A.; Lemieux, G. Isolation of a pure suspension of rat proximal tubules. Am. J. Physiol. 1981, 241, F403–F411. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Non-Diabetic Control Rats | Diabetic Rats | ADORA3 Antagonist-Treated Diabetic Rats | |
|---|---|---|---|
| Body Weight (gr) | 483 ± 56 | 257 ± 70 * | 192 ± 54 * |
| Glycaemia (mg/dL) | 115 ± 14 | 542 ± 82 * | 522 ± 80 * |
| Relative kidney weight (g/Kg) | 4.76 ± 0.99 | 9.83 ± 0.63 * | 8.44 ± 1.9 * |
| Proteinuria (mg/mg creatinine) | 1.1 ± 0.2 | 5.0 ± 0.4 * | 1.93 ± 0.3 # |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garrido, W.; Jara, C.; Torres, A.; Suarez, R.; Cappelli, C.; Oyarzún, C.; Quezada, C.; San Martín, R. Blockade of the Adenosine A3 Receptor Attenuates Caspase 1 Activation in Renal Tubule Epithelial Cells and Decreases Interleukins IL-1β and IL-18 in Diabetic Rats. Int. J. Mol. Sci. 2019, 20, 4531. https://doi.org/10.3390/ijms20184531
Garrido W, Jara C, Torres A, Suarez R, Cappelli C, Oyarzún C, Quezada C, San Martín R. Blockade of the Adenosine A3 Receptor Attenuates Caspase 1 Activation in Renal Tubule Epithelial Cells and Decreases Interleukins IL-1β and IL-18 in Diabetic Rats. International Journal of Molecular Sciences. 2019; 20(18):4531. https://doi.org/10.3390/ijms20184531
Chicago/Turabian StyleGarrido, Wallys, Claudia Jara, Angelo Torres, Raibel Suarez, Claudio Cappelli, Carlos Oyarzún, Claudia Quezada, and Rody San Martín. 2019. "Blockade of the Adenosine A3 Receptor Attenuates Caspase 1 Activation in Renal Tubule Epithelial Cells and Decreases Interleukins IL-1β and IL-18 in Diabetic Rats" International Journal of Molecular Sciences 20, no. 18: 4531. https://doi.org/10.3390/ijms20184531