Microtubules: A Key to Understand and Correct Neuronal Defects in CDKL5 Deficiency Disorder?

Department of Biotechnology and Life Sciences, (DBSV), University of Insubria, Via Manara 7, 21052 Busto Arsizio (VA), Italy
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2019, 20(17), 4075; https://doi.org/10.3390/ijms20174075
Submission received: 25 July 2019
/
Revised: 14 August 2019
/
Accepted: 19 August 2019
/
Published: 21 August 2019
(This article belongs to the Special Issue Molecular Research on Rett Syndrome and Related Disorders: From the Past Towards the Future)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:CDKL5 deficiency disorder (CDD) is a severe neurodevelopmental encephalopathy caused by mutations in the X-linked CDKL5 gene that encodes a serine/threonine kinase. CDD is characterised by the early onset of seizures and impaired cognitive and motor skills. Loss of CDKL5 in vitro and in vivo affects neuronal morphology at early and late stages of maturation, suggesting a link between CDKL5 and the neuronal cytoskeleton. Recently, various microtubule (MT)-binding proteins have been identified as interactors of CDKL5, indicating that its roles converge on regulating MT functioning. MTs are dynamic structures that are important for neuronal morphology, migration and polarity. The delicate control of MT dynamics is fundamental for proper neuronal functions, as evidenced by the fact that aberrant MT dynamics are involved in various neurological disorders. In this review, we highlight the link between CDKL5 and MTs, discussing how CDKL5 deficiency may lead to deranged neuronal functions through aberrant MT dynamics. Finally, we discuss whether the regulation of MT dynamics through microtubule-targeting agents may represent a novel strategy for future pharmacological approaches in the CDD field.
Keywords:
CDKL5; microtubules; neuronal morphology; +TIP; CLIP170; EB1–3; MAP1S; microtubule-targeting agents; pregnenolone1. Introduction
The cyclin-dependent kinase-like 5 (CDKL5) gene is known since 1998, when it was identified in a transcriptional mapping approach to identify disease-causing genes in the Xp22 region [1]. However, the gene caught significant attention only in 2003, when translocations interrupting CDKL5 were identified in two female patients with severe X-linked infantile spasms and intellectual disability [2]. Rapidly afterwards, in 2004, more reports described missense and frame-shift mutations in various positions of CDKL5 in individuals with clinical manifestations, ranging from atypical Rett syndrome (RTT) to autism [3,4,5]. In the following reports, mutations in CDKL5 were commonly identified in patients clinically diagnosed with the early seizure variant of RTT and it was only in 2013 that the pathology associated with CDKL5 deficiency was established as an independent clinical entity referred to as CDKL5 disorder [6] or, lately, as CDKL5 deficiency disorder (CDD). The first clinical manifestation of CDD is normally treatment-resistant seizures that appear within the first three months after birth [7]. Moreover, CDD patients are characterised by developmental delay, intellectual disability, gross motor impairment, strong hypotonia, sleep disturbances, gastrointestinal problems, breathing problems and hand stereotypies. Some of these symptoms overlap with those of RTT, and the main distinction between the two pathologies is the absence of a period of regression, which is among the diagnostic criteria for RTT [8], in patients with CDKL5 mutations.
The clinical manifestations linked to CDKL5 mutations underscore the importance of the encoded serine/threonine kinase for brain functions. Studies aimed at understanding the molecular pathways controlled by CDKL5 and the neuronal and neuroanatomic consequences associated with its deficiency have in the last years provided crucial information of CDKL5 functions. Importantly, very recently, reports from more laboratories converge on a functional link between CDKL5 and microtubules (MTs) [9,10,11,12]. MTs, together with actin and intermediate filaments, constitute a highly dynamic architectural element of neurons and are fundamental for intracellular cargo transport and for the accommodation of structural neuronal changes that underlie synaptic plasticity [13]. Various brain disorders have been linked to mutations in genes that control MT-related processes, and the pharmacological targeting of MTs or the associated proteins are currently being tested in preclinical and clinical studies [14,15].
We will here review the recent findings regarding CDKL5 functions with particular focus on its MT-related functions and the possible translational value of these findings for CDKL5-related disorders.
2. Microtubules
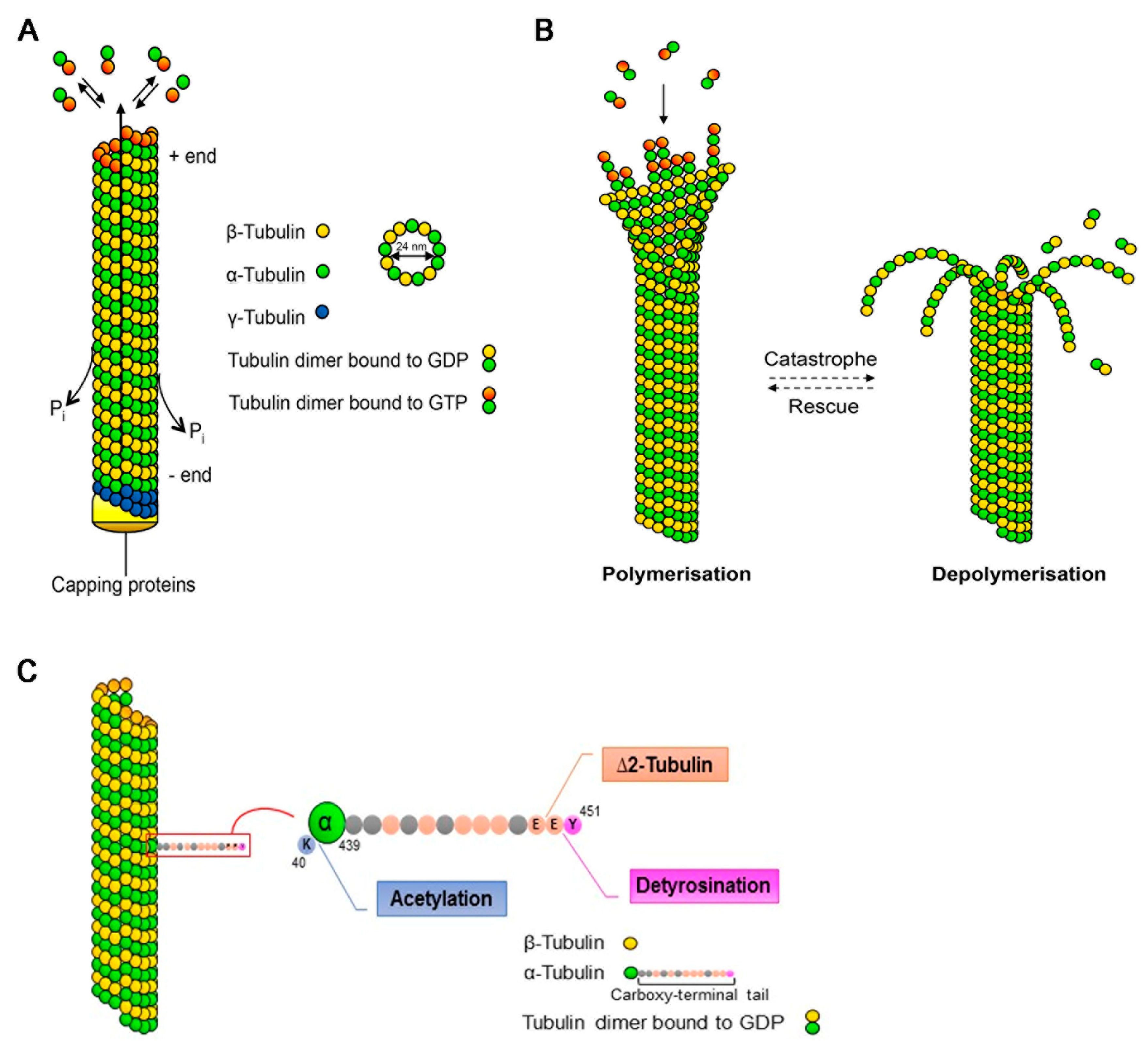
Microtubules are constituted by dimers of α- and β-tubulin, polymerising in a head-to-tail fashion into proto-filaments that assemble laterally to form polarised tubular structures with so-called plus- and minus-ends exposing β- and α-tubulin, respectively [13] (Figure 1A). Assembly requires β-tubulin to be in its guanosine triphosphate (GTP)-bound form; the rapid hydrolysis into guanosine diphosphate (GDP) upon incorporation into the MT lattice generates a GTP-cap at the plus-end, favouring further polymerisation. In most cells, MTs are nucleated from the MT-organising centre (MTOC) with the minus-end of the MTs being embedded and stabilised in the γ-tubulin ring and the plus-ends radiating into the cytoplasm. Neuronal MTs, however, are rapidly released by MT-severing proteins and then transported into the neurites as short polymers. Further, in neurons, MT nucleation can also occur independently of the MTOC [16], with the minus-ends being capped and stabilised by specific proteins. The highly-polarised nature of neurons is mirrored in the organisation of the MT array: in the axon, MTs are uniformly oriented with the plus-ends facing the tip of the axon, while in dendrites they are of mixed polarity with both minus- and plus-ends pointing distally.
MTs are highly dynamic structures that undergo cycles of rapid growth and disassembly in a process defined as dynamic instability [13] (Figure 1B). The switch from growth to disassembly (or shrinkage) is called “catastrophe”, while the reverse is termed “rescue”. This dynamic instability, which occurs mainly at the MT plus-ends, provides a rapid reorganisation of the cytoskeleton that is necessary for many cellular functions, such as cell division and migration and formation of cell polarity. Various factors participate in the control of dynamic instability such as the fine regulation of MT-associated proteins, among which the plus-end tracking proteins (+TIPs) play a fundamental role [17], and the post-translational modifications (PTMs) of α- and β-tubulin that constitute the so-called “tubulin code” [18].
Tubulin PTMs present different allocations on the α/β-tubulin heterodimer with respect to their position on the MT lattice: detyrosination and deglutamylation take place on the C-terminal tail of tubulin that projects away from the MT lattice and provides interaction sites for the MT-associated proteins. Conversely, acetylation of α-tubulin on lysine 40 is found on the inside of the MT lumen (Figure 1C) [18]. Newly-synthesised α-tubulin generally contains a C-terminal tyrosine residue that undergoes enzymatic cyclic removal and re-addition. Detyrosinated MTs are normally considered stable and long-lived, even if the modification per se does not influence MT stability but rather alters the anchorage point for specific proteins. The irreversible removal of the penultimate glutamate from the primary sequence of α-tubulin, generating ∆2-tubulin, prevents α-tubulin re-tyrosination and represents a hallmark of stable MTs. Acetylation of α-tubulin on lysine 40 that, as mentioned, is located within the lumen of the MT lattice is synonymous of stable MTs. Although the causal link between tubulin PTMs and MT stability remains largely enigmatic, the consequences of their disruption can be fatal, as illustrated by animal models carrying inactivating mutations in genes encoding the modifying enzymes [19].
As mentioned, dynamic instability is largely influenced by a plethora of proteins that bind MTs. Classical MT-associated proteins (MAPs) constitute an important group of proteins that bind along the MT lattice, influencing its polymerisation, stability and bundling. MAP1 family proteins, such as MAP1A, MAP1B and MAP1S, are high molecular weight complexes formed of heavy and light chains (LC) [20]. The capacity of these proteins to bind both MTs and actin suggests that they might simultaneously bind and cross-link these two cytoskeletal components. The MAP2 family includes MAP2 and Tau that accumulate in neuronal dendrites and axons, respectively, where they promote MT stability [21].
The +TIP family comprises another large group of diverse proteins that share the common feature of associating with the growing MT plus-end, where they control MT dynamics and functioning [22]. The plus-end tracking behaviour of these proteins can be visualised in live-imaging studies where the binding of the fluorescently tagged proteins to the growing MTs appears as comet-like dashes [23]. The end-binding proteins (EBs) form the central hub of the +TIPs; indeed, they provide the attachment site for other +TIPs thanks to their autonomous binding to MT plus-ends [24]. The EB family comprises EB1–3 that bind MTs through their calponin homology (CH) domain in the Ν-terminus, while they engage in specific protein interactions through C-terminal regions. The cytoskeleton-associated protein Gly-rich (CAP-Gly) domain proteins, including the cytoplasmic linker proteins (CLIP) CLIP170, CLIP115, and p150-Glued, constitute another important group of +TIPs that bind MT plus-ends indirectly via EBs but also by directly recognising the C-terminal EEY/F (glutamate-glutamate-tyrosine/phenylalanine) motif of α-tubulin [19,25,26]. Consistently, detyrosinated α-tubulin is deprived of the binding site for the CAP-Gly proteins. Another large group of +TIPs, including proteins like the CLIP-associated proteins (CLASP), the tumour suppressor adenomatousis polyposis coli protein (APC), p140Cap, and some kinesins (KIF), is characterised by an SxIP (serine-any amino acid-isoleucine-proline) motif through which they bind the EBs [17].
Functionally, the +TIPs can affect MT dynamics in opposing ways with the EBs suppressing the frequency of MT catastrophes and the CLIPs promoting MT rescue [22]. The +TIPs play an essential role in MT-related functions by mediating reciprocal interactions between the MT plus-ends and the cell cortex, where several of these proteins interact directly or indirectly with the actin cytoskeleton. This allows a subset of MTs to be selectively stabilised at specific sites under the cell membrane creating tracks for the delivery of proteins to the cell periphery and pulling forces on the MT network. In this way, the +TIPs are involved in various cellular processes including mitosis, cell migration and morphology [27]. Regarding neurons, various stages of neuronal development such as neuronal migration, axonal growth and dendritic spine maturation are strictly controlled by the +TIPs.
The importance of the proper regulation of the MT network for neuronal functions has been demonstrated through the pharmacological manipulation of MT dynamics. Indeed, axon formation can be artificially induced treating unpolarised neurons with the MT-stabilising drug, taxol [28]. On the other hand, both dendritic outgrowth and the maintenance of mushroom-headed spines were reduced through the inhibition of MT assembly [29,30]. Further, mutations in various genes encoding proteins that regulate MT dynamics can cause defects in neuronal migration and connectivity and have been linked to neurodevelopmental disorders characterised by intellectual disability and autistic features [31].
As mentioned, several links have recently been established between CDKL5 and MTs. Indeed, CDKL5 has recently been found to interact with the two +TIPs, CLIP170 and EB2 [9,10,11]. The latter was identified through a chemical genetic approach as a CDKL5 substrate and was thoroughly characterised as a bona fide phosphorylation target of CDKL5 in vitro and in vivo; the functional consequences of this event are still unknown, however [11], and neuronal functions of EB2 have not been described so far. CLIP170 was related functionally to CDKL5 through the identification of IQ motif containing GTPase activating protein 1 (IQGAP1) as a novel CDKL5 interactor [9]. IQGAP1 is an actin-binding protein that binds CLIP170 in a Rac1-dependent manner, facilitating the transient capture of MTs at specific cortical sites under the cell membrane [32,33]. Further, MAP1S, the less characterised member of the MAP1 family proteins, was identified as a substrate of CDKL5 through two different approaches providing strong support of a functional role of this association [11,12].
Since the discovery of the first patients with CDKL5 mutations, CDKL5 functions have been studied in vitro and in vivo in proliferating cells, primary neurons, in mouse models carrying inactivating deletions of the Cdkl5-gene and in human induced pluripotent stem cells (iPSC)-derived neurons [34]. The overall picture emerging from these studies indicates that CDKL5 deficiency impacts cell proliferation, neuronal migration, and various aspects of neuronal morphology that are all processes that are strictly influenced by MT dynamics. These findings, demonstrating that CDKL5 functions converge on MT dynamics, may, therefore, provide a key to understand the mechanisms underlying CDD in more details.
3. Neuronal CDKL5 Related Defects
3.1. Axon Formation
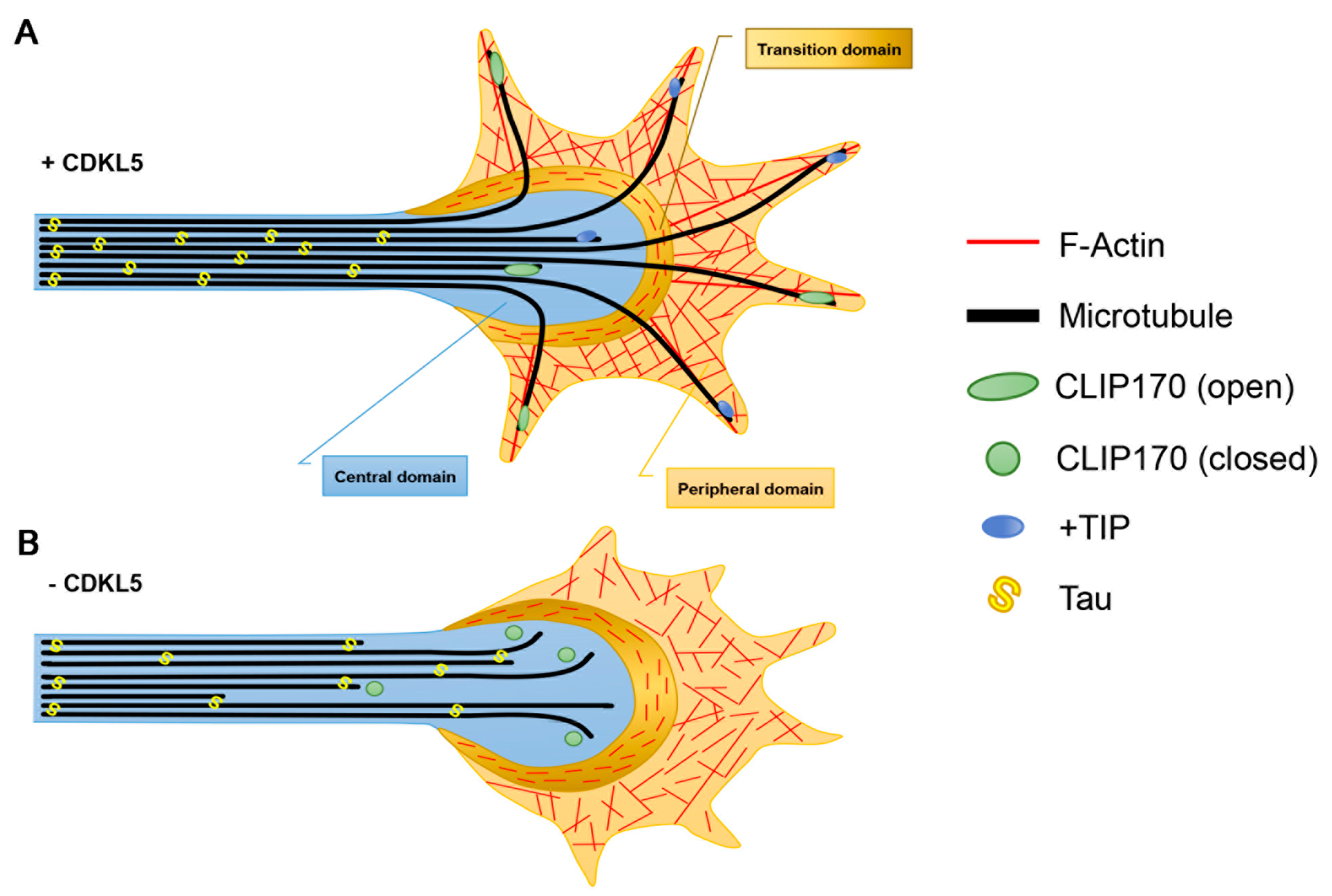
In primary low-density cultures of hippocampal neurons, CDKL5 controls axon specification and outgrowth [35,36]. Axon elongation relies on the formation of stable MTs in the axonal shaft. These MTs form bundles covered with the axon specific MAP, Tau, and are organised in a polarised fashion, with the plus-ends oriented towards the peripheral tip. Such MT organisation generates tracks that facilitate the transport of proteins and organelles, thus providing the building blocks for new axonal segments. The axonal tip is characterised by a fan-shaped, motile structure, the growth cone, that probes the extracellular environment. In this way axon elongation and steering is directed in a process that depends on the complex remodelling and reorganisation of MTs and actin (Figure 2). The growth cone comprises the central region, enriched in MTs, and the peripheral domain, which is mainly composed of filamentous actin (F-actin); the two regions are separated by the transition zone in which actin arcs form a barrier against the extension of MTs into the peripheral zone [37]. Few MTs are capable of escaping the central domain and reach the growth cone periphery where they, thanks to the +TIPs, can couple to F-actin and associate with specific sites at the growth cone cortex. In particular, the +TIP CLIP170 accumulates in the axonal growth cone by binding to tyrosinated tubulin [19,38]. Interfering with CLIP170 expression impairs axon formation in primary neurons. This is accompanied by a reduced capacity of MTs to penetrate the actin arcs and protrude into the peripheral domain [39]. CDKL5 deficiency reduces the association of CLIP170 to MTs in proliferating cells and in the axonal growth cone of Cdkl5-KO neurons, which in both cases is accompanied by altered MT dynamics [9,40]. The binding of CLIP170 to MTs occurs when the protein is in its open active conformation whereas the intramolecular interaction between its terminal CAP-Gly domain and zinc-knuckle motifs causes a closed inactive conformation [41]. Interestingly, the neurosteroid pregnenolone (PREG), which induces the open conformation of CLIP170 through its direct binding to the protein [42], is capable of normalising the MT-binding of CLIP170 and restore axon specification and elongation in CDKL5 deficient neurons [9]. It thus seems likely that CDKL5 is involved in maintaining CLIP170 in its active conformation. The switching between the open and closed conformation of CLIP170 is known to depend on specific phosphorylation events. Whether CDKL5 regulates CLIP170 in a phosphorylation-dependent manner is still not known.
In the past, CDKL5 was proposed to affect axon formation through its interaction with Shootin1 [35]. Shootin1 is a key determinant for axon specification [43]; it was found to act as a clutch molecule that produces the traction force for axon outgrowth by coupling the retrograde flow of F-actin and cell adhesions [44]. Considering the novel link with CLIP170 it, thus, appears that CDKL5 may influence axon formation in different ways.
3.2. Dendritic Arborisation
CDKL5 deficiency was first shown, through silencing studies, to influence dendritic arborisation in vitro in primary cultures of rat cortical neurons and in vivo in migrating neurons derived from silenced neural progenitor cells [45]. Such defect has later been confirmed in cortical and hippocampal pyramidal neurons in Cdkl5-KO brains [46,47,48] and reproduced in vitro in Cdkl5-KO primary hippocampal neurons [36,40]. The penetrance of the phenotype in vitro may depend on the experimental conditions since contrasting data have been reported [11].
The control of CDKL5 on dendrite morphology was found to occur through Rac1, which is a critical regulator of actin remodelling. Recombinant CDKL5 was capable of pulling out Rac1 from neuronal lysates, an interaction that was further stimulated upon prior neuronal stimulation with brain-derived neurotrophic factor (Bdnf). Further, Bdnf activation of Rac1 was impaired in CDKL5 silenced cells, thus placing Rac1 downstream the kinase [45]. The exact mechanism through which CDKL5 activates Rac1 is unknown; however, it may be relevant to consider that CDKL5 controls the stability of the ternary IQGAP1/CLIP170/Rac1 complex [9]. IQGAP1 is widely implicated in Rac1/cdc42 signalling [49]. Indeed, IQGAP1 maintains the two small GTPases in their active GTP-bound state and acts as a scaffolding platform for Rac1/cdc42 signalling by recruiting their downstream effector proteins. Further, CLIP170 and IQGAP1 were reported to cooperate in controlling dendrite morphology, apparently by bridging MTs and the actin cytoskeleton [50]. Reduced CLIP170 or IQGAP1 levels led to the formation of a less complex dendritic arbor, a phenotype that resembles the one linked to CDKL5 deficiency and that could be prevented through the pharmacological stabilisation of F-actin. CDKL5 may, thus, control the cytoskeleton dynamics involved in dendrite morphology by allowing the formation of the protein complex mediating Rac1 activation and bridging the MT and actin networks.
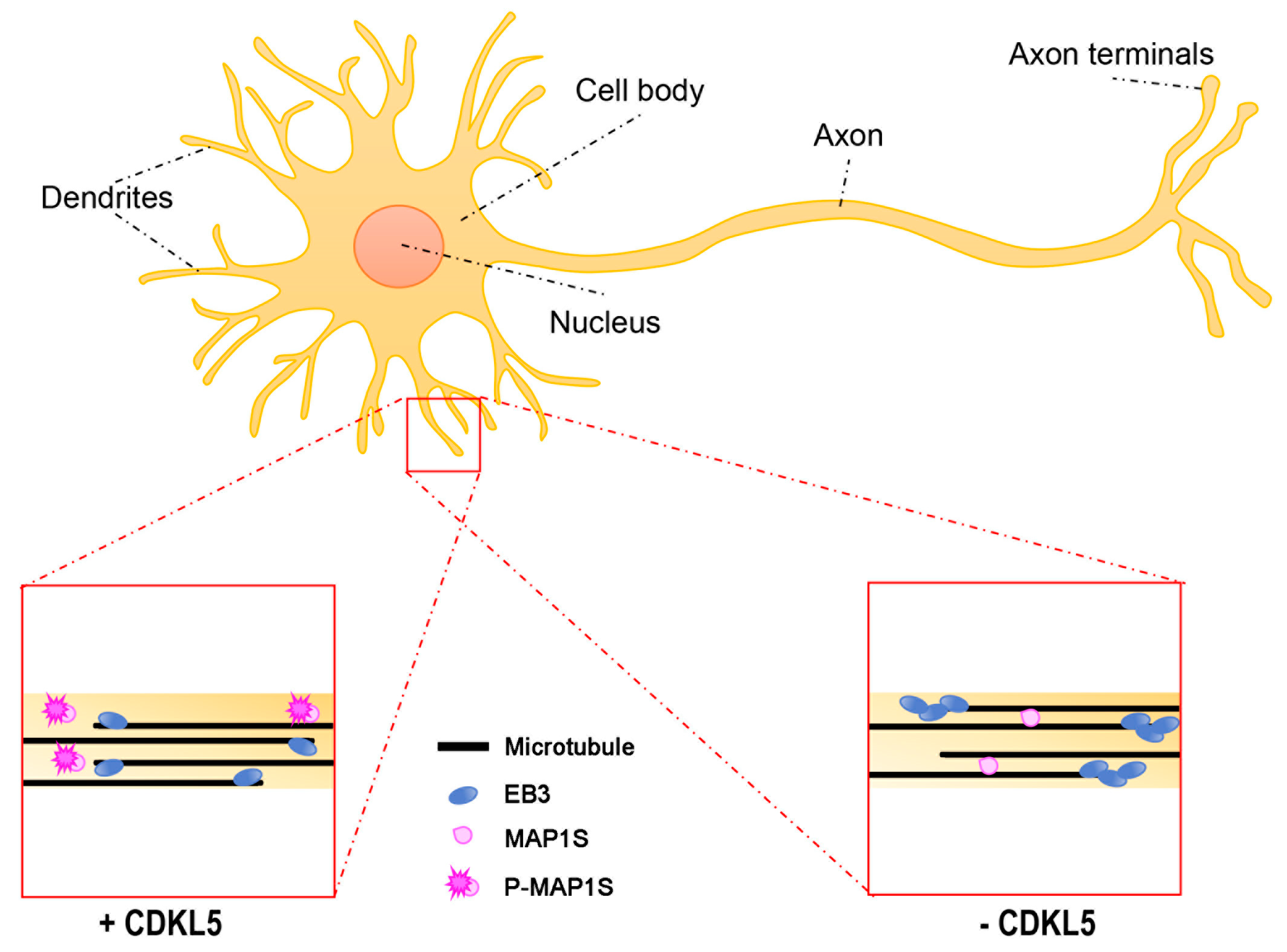
Another possible way in which CDKL5 regulates MT dynamics during dendritic arborisation may be represented by the phosphorylation on MAP1S, recently identified as a bona fide substrate of CDKL5 [11,12] (Figure 3). When phosphorylated on two serines (Ser786 and Ser812), located within the LC, MAP1S LCs have reduced affinity for MTs and appear to be more soluble. Interestingly, overexpression of phospho-defective MAP1S LCs in neurons forced the dendritic tips to settle in an aberrant looped conformation. In line with the capacity of MAP1 family members to increase MT stability [51], functional studies suggested that the increased binding of MAP1S to MTs in the absence of CDKL5 leads to less dynamic MT plus-ends. Indeed, EB3-GFP (green fluorescent protein) comets, the fluorescence of which over time is a measure of MT dynamicity, are longer lived in Cdkl5-KO neurons, but are restored to normal life-times upon MAP1S silencing [11].
3.3. Dendritic Spines
Various studies in vitro and in vivo converge on a role of CDKL5 in controlling dendritic spine morphology and excitatory functions. Neurons devoid of CDKL5 are characterised by an increased number of filopodia-like protrusions and a reduced number of mushroom-shaped spines [52,53]. The enlarged spine heads, which distinguish mushroom-shaped spines, correlate with increased synaptic strength. Various neurologic conditions characterised by cognitive dysfunctions and autistic features have been linked to altered density and shape of dendritic spines [54]. CDKL5 is readily detectable in dendritic spines, where its synaptic accumulation is regulated both by the dendritic targeting of its mRNA, which undergoes local activity-dependent translation, and through its interaction with post-synaptic density protein 95 (PSD95) [52,55,56]. CDKL5 is, thus, likely to influence directly various proteins involved in excitatory neurotransmission. Accordingly, CDKL5 affects the expression or phosphorylation of the post-synaptic markers PSD95, Homer, the α-amino-3-hydroxy-5-methylisozasole-4-propionic acid (AMPA)-receptor subunit GluA2, and NGL1 [52,56,57,58]. So far, we have very little information whether the neuroanatomical and molecular alterations in CDD mouse models are present also in the patients. However, reduced membrane levels of GluA2, which have been observed in vitro in neurons silenced for CDKL5 expression and in vivo in the hippocampus and perirhinal cortex of two mouse models of CDD, were recently confirmed in post-mortem brains from two CDD patients [58,59,60], suggesting the relevance of at least some CDKL5-related defects for the human pathology.
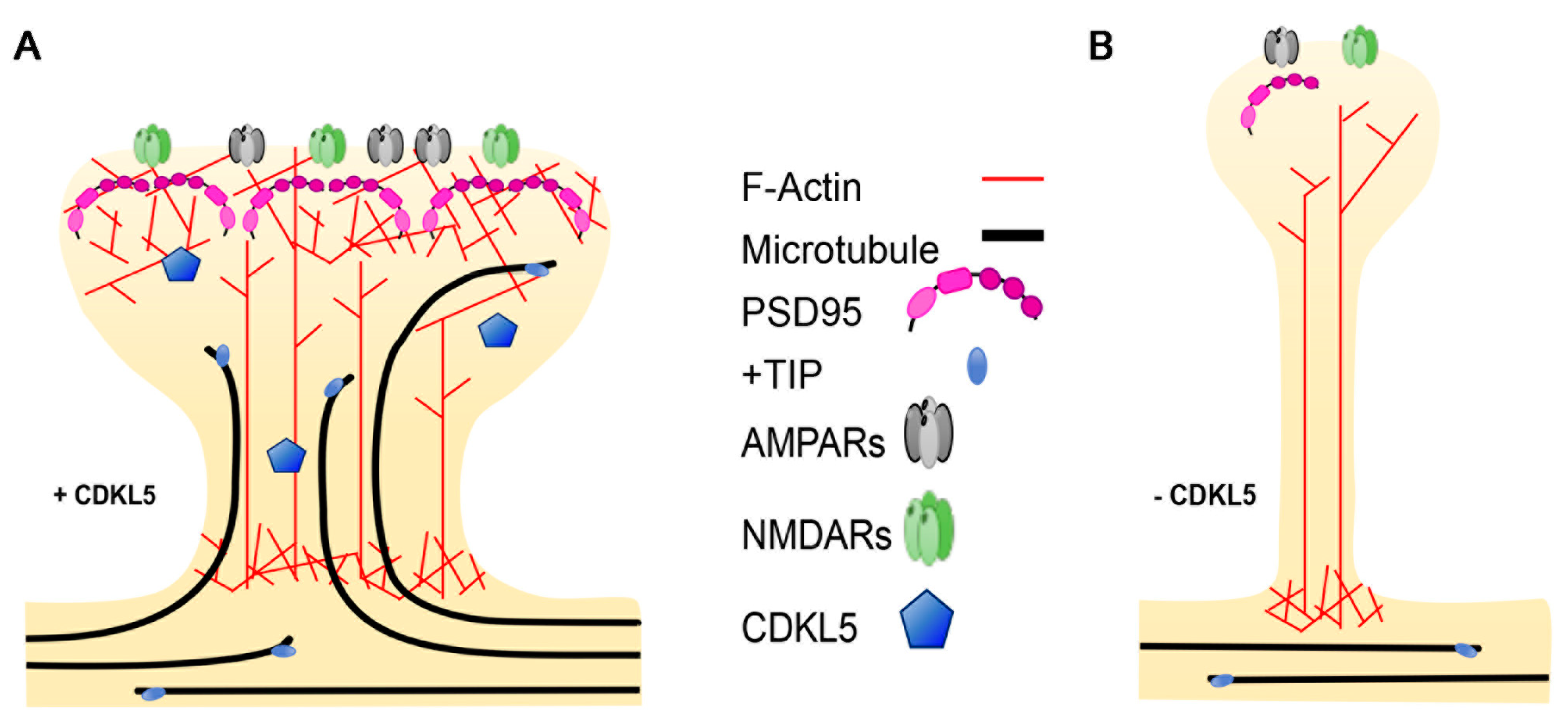
Plasticity-related changes in spine morphology are widely known to rely on remodelling of the actin cytoskeleton, which is highly enriched in dendritic protrusions [61]. MTs were for many years ignored as an integral part of dendritic spines; however, live imaging studies of fluorescently-tagged EB3, marking dynamic MTs, combined with other techniques, have allowed demonstrating that MTs do actually polymerise into dendritic spines in a rapid and transient manner [30,62,63] (Figure 4). The incursion of MTs into the spines is activity-dependent and contributes to the formation of mature dendritic spines. Actually, nocodazole-mediated inhibition of dynamic MTs impedes the Bdnf-induced structural changes that leads to the maturation of filopodia-like protrusions into mushroom-shaped spines [63]. The +TIP EB3 plays an active role in MT-dependent structural plasticity; actually, its silencing leads to a reduced number of mushroom-shaped spines. Paralleling the importance of a cross-talk between MTs and the actin cytoskeleton in the axonal growth cone, EB3 was found to provide a link to the actin cytoskeleton in the spines. Indeed, interactions between MT-bound EB3 with p140Cap and the actin stabilising factor cortactin is likely to create local signalling events that allow MT-actin communication in “activated” spines [30]. Interestingly, the actin–MT cooperation seems, in turn, to be enhanced by neuronal network activation; indeed, very recent data suggest that MT invasion occurs in specific spines through N-methyl-D-aspartate (NMDA)-receptor-mediated calcium influx that leads to actin remodelling at the base of the spine, thus allowing the targeting of the MTs [64].
Regarding a possible role of CDKL5 in regulating spine morphogenesis through MT-related events, it is tempting to speculate that more +TIPs may be involved in regulating spine morphogenesis and functions creating other distinct links between MTs and the actin cytoskeleton. This would allow an expansion in the repertoire of possible signalling events underlying structural plasticity. While EB2 is broadly expressed in the neuronal soma, its levels are very low in the post-synaptic compartment [65]. Accordingly, CDKL5-dependent phosphorylation of EB2 on serine 223 can readily be detected in the proximal dendrites of patient iPSC-derived neurons [11]. The presence of CLIP170 in dendritic spines has so far not been reported. However, IQGAP1, which interacts with both CDKL5 and CLIP170, is present in spines [66], where it regulates spine morphogenesis [67], and Iqgap1-KO mice display cognitive defects and impaired long-term potentiation (LTP) [68]. IQGAP1 interacts with various post-synaptic proteins such as the AMPA-receptor GluA4, PSD95 and the NMDA-receptor complex [68,69]; it is, thus, intriguing to speculate that its interaction with CLIP170 might serve a role in regulating spine formation and functioning. Of further interest, disks large homolog 5 (DLG5), which was identified as a cellular substrate of CDKL5 [12], is an important regulator of dendritic spine formation [70]. DLG5 is member of the membrane-associated guanylate kinase (MAGUK) proteins and appears to control spine formation by facilitating the localisation of N-cadherin to the cell membrane. A possible link between DLG5 and MTs can be hypothesised based on its interaction with MARK3 (MT affinity regulating kinase 3), a kinase that controls cell polarity through the phosphorylation of specific MAPs, thus regulating their affinity for MTs [71].
Considering that MTs constitute fundamental tracks for the motor-based transport of cargoes, their invasion into dendritic spines is likely to provide a way of transporting cargoes into and out of these structures. The nature of which cargoes enter spines through MTs is still not clear. However, synaptotagmin has been found to be transported in a kinesin-dependent manner into dendritic spines [72]. Further studies will be relevant to analyse the contribution of altered MT dynamics to CDKL5-related spine defects and which cargoes might be influenced by CDKL5 deficiency.
4. Non-Neuronal CDKL5-Related Defects
The role of CDKL5 in regulating MT dynamics is consistent with our previous report that described CDKL5 as a centrosomal protein [10]. The centrosome is a central component of the cytoskeletal structure and consists of a scaffold core containing a large number of proteins, including γ-tubulin and the γ-tubulin ring complex (γ-TuRC) that typically surrounds a pair of cylindrical centrioles enclosed in the amorphous pericentriolar material (PCM) [73]. The centrosome is the primary MT-organising centre of the cells and, therefore, participates in fundamental cellular functions, such as cell motility, migration, polarity [74,75] and in the assembly and organisation of the mitotic spindle [76,77]. CDKL5 silencing causes an increase in the number of defective multipolar spindles, which is associated with polyploidisation and centrosome accumulation [10]. It is interesting to note that CDKL5 was recently found to interact with and phosphorylate the centrosomal protein CEP131 on serine 35 [12]. The functional role of this phosphorylation event was not further explored; however, silencing of CEP131 has been reported to cause centriole amplification and chromosomal instability [78], a phenotype that overlaps the one caused by CDKL5 deficiency [10]. Furthermore, CDKL5 and CEP131 share a common role in ciliogenesis [79,80]. The primary cilium is a eukaryotic organelle that is present on most quiescent and differentiated cells; it originates from a centrosome-derived structure, the basal body, and is composed by a central MT structure, the axoneme. The primary cilium acts as a cellular antenna that transmits signals from the extracellular environment to the inner site of the cell, thereby contributing to the regulation of numerous cellular processes such as proliferation, differentiation, migration and cell polarity [81]. Interestingly, defects in cilia morphogenesis and functioning have been associated with a large list of heterogeneous genetic disorders, known as ciliopathies [82]. Among the various clinical manifestations linked to ciliary dysfunctions, some share an overlap with those of CDD such as developmental delay, epilepsy, cognitive dysfunction and aberrant respiration. Cilium length appears to be influenced by CDKL5 levels [79,83]; a further understanding of how CDKL5 dysfunction impacts ciliary functions and the contribution of CEP131 and MT dynamics may pave the way for new perspectives in the CDKL5 field.
5. Possible Functional Outcome of Specific Pathogenic CDKL5 Mutations
The above studies linking CDKL5 to MT-related functions have been addressed in cells or tissues silenced for CDKL5 or with no expression of the protein. This reflects well the current view that CDD is caused by the absence of functional CDKL5 or by the expression of hypomorphic derivatives. Indeed, CDD-causing mutations in CDKL5 include missense mutations, nonsense, frame-shift and splicing variants, and large genomic deletions [84]. The pathogenic missense mutations, which have been identified so far, fall almost exclusively within the catalytic domain of the protein. Functional studies based on the exogenous expression of mutated CDKL5 derivatives suggest that amino acid substitutions within the kinase domain impact negatively its catalytic activity [79,85,86,87]. Although these studies have highlighted the impact of missense mutations on CDKL5 functioning, it needs to be underlined that only very few of these mutations have been studied; we therefore cannot exclude that other missense mutations may generate hypermorphic CDKL5 derivatives. Regarding the nonsense and frame-shift variants, nonsense-mediated decay of the mutated mRNA is likely to impede the expression of the encoded protein, thus acting as hypomorphic or null mutations [84,88,89].
The existing animal models of CDD have addressed the consequences of the absence of CDKL5 either in the whole body or in distinct brain areas [46,47,48,90]. Recently, studies were performed in a mouse line expressing the pathogenic R59X allele, but since this causes the absence of CDKL5, it can be considered a functional null [60,91]. Although the generation of CDD mouse models has been extremely useful to deeply understand the roles of CDKL5, an important aspect that still remains to be addressed is how the expression of pathogenic hypomorphic CDKL5 derivatives affects neuronal functions. Hypothetically, such derivatives could behave as dominant negatives and interfere with the normal function of CDKL5 interactors, such as the MT-interacting proteins. Of relevance, CDKL5 localisation and cilium length is influenced by the expression of CDKL5 derivatives containing CDD-causing missense mutations that have not yet been characterised for the catalytic activity [79]. While the field is awaiting such animal models, primary fibroblasts and iPSC-derived neurons obtained from CDD patients will be very informative. Finally, it must be remembered that large genomic duplications, including CDKL5, have been identified in individuals with varying degrees of macrocephaly and learning disabilities [92], suggesting the relevance of analysing the effect of increased CDKL5 levels on MT-related functions.
6. Therapeutic Relevance of CDKL5-Dependent MT Defects
Alterations of the MT cytoskeleton have been linked to neurodegenerative and neurodevelopmental disorders such as schizophrenia and autism and major depression disorder [15,93]. Efforts have, therefore, been put into the development of therapeutic strategies targeting the neuronal cytoskeleton for these disorders [14,94]. The pharmacological stabilisation of MTs with the consequent block of mitosis has for decades been used for cancer treatment; the possibility of repositioning these drugs has been considered for brain disorders. MT-stabilising drugs were initially considered for neurodegenerative disorders that are characterised by compromised tau functions and altered MT structure and axonal transport [94]. Taxol (paclitaxel) promotes MT stability by binding within the MT lumen and increasing the lateral interactions between adjacent proto-filaments [95]. When considering the use of MT-stabilising drugs for brain disorders, it is fundamental to consider their capacity to pass the blood–brain barrier and their possible neuropathy side effects. While proof-of-principle studies supported the beneficial effect of taxol-mediated MT stabilisation on a mouse model presenting tau pathology in spinal motor neurons [96], this drug has a limited bioavailability in brain since it does not cross the blood–brain barrier. As taxol, Epothilone D (EpoD) binds and stabilises MTs, but it has the advantage of being brain penetrant, thus holding a stronger therapeutic potential for brain disorders [97]. EpoD has been tested in animal models of tauopathies where doses much lower (1/30) than those used in cancer trials were found to be effective in stabilising MTs and to be capable of reducing cognitive deficits present in these models [98,99]. While EpoD and other drugs with similar MT-stabilising activities may possess interesting therapeutic potentials for patients with neurodegenerative diseases, their clinical application for young patient groups, as for example CDD, is more difficult to imagine. Drugs that bind MTs directly will impact MT stability in a non-specific manner and may, thus, have broad and non-advantageous effects on neurodevelopmental processes that are still ongoing in young patients. The use of drugs that target MT-binding proteins may act more specifically and, therefore, represent valid therapeutic options. An interesting molecule is represented by NAP/davunetide, an eight amino acid peptide that is derived from the activity-dependent neuroprotective protein (ADNP) [100]. ADNP is a versatile protein interacting with SWI/SNF chromatin remodelling complexes and with the +TIPs EB1 and EB3 [101,102]. Mutations in ADNP have been linked to autism and intellectual disability, and Adnp haploinsufficiency in mice is linked to altered axonal transport and dendritic spine alterations [103]. The NAP peptide interacts with the EB-proteins and promotes axonal transport and MT stability [104]. In pre-clinical studies in mouse models of various neurodegenerative diseases NAP shows neuroprotection and improves cognitive defects [105]. In clinical studies, intra-nasal administration of NAP led to improved cognitive skills in Alzheimer’s disease patients [106] and daily activities in schizophrenic patients [107].
The possibility of targeting of proteins that have been functionally linked to CDKL5 might hold an even higher therapeutic potential for CDD. Interestingly, the neuroactive steroid PREG was found to directly bind CLIP170, inducing its open active conformation, as well as MAP2 [108]. Notably, the treatment of CDKL5-silenced cells with PREG was capable of restoring the MT association of GFP-CLIP170 comets and neuronal defects linked to CDKL5 deficiency [9]. Treatment with PREG is known to enhance memory and cognition in rodents [109]. Further, clinical testing in humans indicated potential anti-depressive and anti-psychotic effects and a positive safety profile [110,111]. However, the clinical application of PREG is limited due to its short half-life and its conversion into other neuroactive steroids that altogether possess a broad range of activities acting at the genomic level and with numerous membrane-bound receptors [108]. The development and testing of synthetic derivatives with improved stability and that cannot be metabolised into the downstream steroids might represent a possibility for CDD. Of note, MAPREG is a synthetic PREG derivative that has anti-depressant activities in rat models of depression [112] and in the primate-like tree-shrew [113]. Like PREG, MAPREG/MAP4343 promotes MT dynamics apparently through its binding to MAP2. Through further studies it might be relevant to analyse the therapeutic potential of this or similar PREG derivatives on CDKL5-deficient neurons.
To conclude, the last findings of CDKL5 functions converging on MT dynamics are likely to be of great relevance not only for a deeper understanding of the neuronal dysfunctions linked to CDKL5 deficiency but also for the development of disease-modifying pharmacological approaches. Importantly, they may also lead to the identification of clinical diagnostic biomarkers. However, before considering MT-targeting drugs for clinical applications for CDD, we still need to understand in more detail how precisely CDKL5 deficiency affects MT dynamics.
Author Contributions
C.K.N. and I.B., writing—original draft preparation; R.D.R., reviewing, editing, figure preparation.
Funding
This work was supported by funding from the Telethon Foundation grant (GGP15098) to CKN.
Acknowledgments
We thank the Italian parents’ association l’Albero di Greta for the financial support of the laboratory.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| +TIP | plus-end tracking protein |
| ADNP | activity-dependent neuroprotective protein |
| AMPA | α-amino-3-hydroxy-5-methylisozasole-4-propionic acid |
| APC | adenomatousis polyposis coli |
| Bdnf | brain-derived neurotrophic factor |
| CAP-Gly | cytoskeleton-associtated protein Gly-rich |
| CDD | CDKL5 deficiency disorder |
| CDKL5 | cyclin-dependent kinase-like 5 |
| CH | calponin homology |
| CLASP | CLIP-associated protein |
| CLIP | cytoplasmic linker protein |
| DLG5 | disks large homolog 5 |
| E | glutamate |
| EB | end-binding protein |
| EEY/F | glutamate-glutamate-tyrosine/phenylalanine |
| EpoD | Epothilone D |
| F-actin | filamentous actin |
| γ-TuRC | γ-tubulin ring complex |
| GFP | green fluorescent protein |
| GTP | guanine triphospate |
| GDP | guanine diphosphate |
| IQGAP1 | IQ motif containing GTPase activating protein 1 |
| iPSC | induced pluripotent stem cell |
| K | lysine |
| KIF | kinesin |
| LC | light chain |
| LTP | long-term potentiation |
| MAGUK | membrane-associated guanylate kinase |
| MAP | microtubule associated protein |
| MARK3 | MT affinity regulating kinase 3 |
| MT | microtubule |
| MTOC | microtubule organising centre |
| NMDA | N-methyl-D-aspartate |
| PCM | pericentriolar material |
| PREG | pregnenolone |
| PSD95 | post-synaptic density protein 95 |
| PTM | post-translational modification |
| RTT | Rett syndrome |
| SxIP | serine-any protein-isoleucine-proline |
| Y | tyrosine |
References
- Montini, E.; Andolfi, G.; Caruso, A.; Buchner, G.; Walpole, S.M.; Mariani, M.; Consalez, G.; Trump, D.; Ballabio, A.; Franco, B. Identification and characterization of a novel serine-threonine kinase gene from the Xp22 region. Genomics 1998, 51, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Kalscheuer, V.M.; Tao, J.; Donnelly, A.; Hollway, G.; Schwinger, E.; Kübart, S.; Menzel, C.; Hoeltzenbein, M.; Tommerup, N.; Eyre, H.; et al. Disruption of the serine/threonine kinase 9 gene causes severe X-linked infantile spasms and mental retardation. Am. J. Hum. Genet. 2003, 72, 1401–1411. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Van Esch, H.; Hagedorn-Greiwe, M.; Hoffmann, K.; Moser, B.; Raynaud, M.; Sperner, J.; Fryns, J.-P.; Schwinger, E.; Gécz, J.; et al. Mutations in the X-linked cyclin-dependent kinase-like 5 (CDKL5/STK9) gene are associated with severe neurodevelopmental retardation. Am. J. Hum. Genet. 2004, 75, 1149–1154. [Google Scholar] [CrossRef]
- Weaving, L.S.; Christodoulou, J.; Williamson, S.L.; Friend, K.L.; McKenzie, O.L.D.; Archer, H.; Evans, J.; Clarke, A.; Pelka, G.J.; Tam, P.P.L.; et al. Mutations of CDKL5 cause a severe neurodevelopmental disorder with infantile spasms and mental retardation. Am. J. Hum. Genet. 2004, 75, 1079–1093. [Google Scholar] [CrossRef]
- Scala, E.; Ariani, F.; Mari, F.; Caselli, R.; Pescucci, C.; Longo, I.; Meloni, I.; Giachino, D.; Bruttini, M.; Hayek, G.; et al. CDKL5/STK9 is mutated in Rett syndrome variant with infantile spasms. J. Med. Genet. 2005, 42, 103–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fehr, S.; Wilson, M.; Downs, J.; Williams, S.; Murgia, A.; Sartori, S.; Vecchi, M.; Ho, G.; Polli, R.; Psoni, S.; et al. The CDKL5 disorder is an independent clinical entity associated with early-onset encephalopathy. Eur. J. Hum. Genet. 2013, 21, 266–273. [Google Scholar] [CrossRef]
- Olson, H.E.; Demarest, S.T.; Pestana-Knight, E.M.; Swanson, L.C.; Iqbal, S.; Lal, D.; Leonard, H.; Cross, J.H.; Devinsky, O.; Benke, T.A. Cyclin-Dependent Kinase-Like 5 Deficiency Disorder: Clinical Review. Pediatr. Neurol. 2019, 97, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Neul, J.L.; Kaufmann, W.E.; Glaze, D.G.; Christodoulou, J.; Clarke, A.J.; Bahi-Buisson, N.; Leonard, H.; Bailey, M.E.S.; Schanen, N.C.; Zappella, M.; et al. Rett syndrome: Revised diagnostic criteria and nomenclature. Ann. Neurol. 2010, 68, 944–950. [Google Scholar] [CrossRef] [Green Version]
- Barbiero, I.; Peroni, D.; Tramarin, M.; Chandola, C.; Rusconi, L.; Landsberger, N.; Kilstrup-Nielsen, C. The neurosteroid pregnenolone reverts microtubule derangement induced by the loss of a functional CDKL5-IQGAP1 complex. Hum. Mol. Genet. 2017, 26, 3520–3530. [Google Scholar] [CrossRef] [PubMed]
- Barbiero, I.; Valente, D.; Chandola, C.; Magi, F.; Bergo, A.; Monteonofrio, L.; Tramarin, M.; Fazzari, M.; Soddu, S.; Landsberger, N.; et al. CDKL5 localizes at the centrosome and midbody and is required for faithful cell division. Sci. Rep. 2017, 7, 6228. [Google Scholar] [CrossRef]
- Baltussen, L.L.; Negraes, P.D.; Silvestre, M.; Claxton, S.; Moeskops, M.; Christodoulou, E.; Flynn, H.R.; Snijders, A.P.; Muotri, A.R.; Ultanir, S.K. Chemical genetic identification of CDKL5 substrates reveals its role in neuronal microtubule dynamics. EMBO J. 2018, 37, e99763. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, I.M.; Morgan, M.E.; Peltier, J.; Weiland, F.; Gregorczyk, M.; Brown, F.C.; Macartney, T.; Toth, R.; Trost, M.; Rouse, J. Phosphoproteomic screening identifies physiological substrates of the CDKL5 kinase. EMBO J. 2018, 37, e99559. [Google Scholar] [CrossRef] [PubMed]
- Kapitein, L.C.; Hoogenraad, C.C. Building the Neuronal Microtubule Cytoskeleton. Neuron 2015, 87, 492–506. [Google Scholar] [PubMed] [Green Version]
- Varidaki, A.; Hong, Y.; Coffey, E.T. Repositioning Microtubule Stabilizing Drugs for Brain Disorders. Front. Cell. Neurosci. 2018, 12, 226. [Google Scholar] [CrossRef] [PubMed]
- Bonini, S.A.; Mastinu, A.; Ferrari-Toninelli, G.; Memo, M. Potential Role of Microtubule Stabilizing Agents in Neurodevelopmental Disorders. Int. J. Mol. Sci. 2017, 18, 1627. [Google Scholar] [Green Version]
- Stiess, M.; Maghelli, N.; Kapitein, L.C.; Gomis-Rüth, S.; Wilsch-Bräuninger, M.; Hoogenraad, C.C.; Tolić-Nørrelykke, I.M.; Bradke, F. Axon extension occurs independently of centrosomal microtubule nucleation. Science 2010, 327, 704–707. [Google Scholar] [CrossRef]
- van de Willige, D.; Hoogenraad, C.C.; Akhmanova, A. Microtubule plus-end tracking proteins in neuronal development. Cell. Mol. Life Sci. 2016, 73, 2053–2077. [Google Scholar]
- Yu, I.; Garnham, C.P.; Roll-Mecak, A. Writing and Reading the Tubulin Code. J. Biol. Chem. 2015, 290, 17163–17172. [Google Scholar] [CrossRef] [Green Version]
- Erck, C.; Peris, L.; Andrieux, A.; Meissirel, C.; Gruber, A.D.; Vernet, M.; Schweitzer, A.; Saoudi, Y.; Pointu, H.; Bosc, C.; et al. A vital role of tubulin-tyrosine-ligase for neuronal organization. Proc. Natl. Acad. Sci. USA 2005, 102, 7853–7858. [Google Scholar] [Green Version]
- Halpain, S.; Dehmelt, L. The MAP1 family of microtubule-associated proteins. Genome Biol. 2006, 7, 224. [Google Scholar] [CrossRef]
- Dehmelt, L.; Halpain, S. Actin and microtubules in neurite initiation: Are MAPs the missing link? J. Neurobiol. 2004, 58, 18–33. [Google Scholar] [CrossRef]
- Akhmanova, A.; Steinmetz, M.O. Tracking the ends: A dynamic protein network controls the fate of microtubule tips. Nat. Rev. Mol. Cell Biol. 2008, 9, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Perez, F.; Diamantopoulos, G.S.; Stalder, R.; Kreis, T.E. CLIP-170 highlights growing microtubule ends in vivo. Cell 1999, 96, 517–527. [Google Scholar] [CrossRef]
- Mustyatsa, V.V.; Boyakhchyan, A.V.; Ataullakhanov, F.I.; Gudimchuk, N.B. EB-Family Proteins: Functions and Microtubule Interaction Mechanisms. Biochem. Mosc. 2017, 82, 791–802. [Google Scholar] [CrossRef] [PubMed]
- Peris, L.; Thery, M.; Fauré, J.; Saoudi, Y.; Lafanechère, L.; Chilton, J.K.; Gordon-Weeks, P.; Galjart, N.; Bornens, M.; Wordeman, L.; et al. Tubulin tyrosination is a major factor affecting the recruitment of CAP-Gly proteins at microtubule plus ends. J. Cell Biol. 2006, 174, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Bieling, P.; Kandels-Lewis, S.; Telley, I.A.; van Dijk, J.; Janke, C.; Surrey, T. CLIP-170 tracks growing microtubule ends by dynamically recognizing composite EB1/tubulin-binding sites. J. Cell Biol. 2008, 183, 1223–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, N.; Draviam, V.M. Microtubule plus-ends within a mitotic cell are “moving platforms” with anchoring, signalling and force-coupling roles. Open Biol. 2012, 2, 120132. [Google Scholar] [CrossRef]
- Witte, H.; Neukirchen, D.; Bradke, F. Microtubule stabilization specifies initial neuronal polarization. J. Cell Biol. 2008, 180, 619–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharp, D.J.; Yu, W.; Baas, P.W. Transport of dendritic microtubules establishes their nonuniform polarity orientation. J. Cell Biol. 1995, 130, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Jaworski, J.; Kapitein, L.C.; Gouveia, S.M.; Dortland, B.R.; Wulf, P.S.; Grigoriev, I.; Camera, P.; Spangler, S.A.; Di Stefano, P.; Demmers, J.; et al. Dynamic microtubules regulate dendritic spine morphology and synaptic plasticity. Neuron 2009, 61, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Lasser, M.; Tiber, J.; Lowery, L.A. The Role of the Microtubule Cytoskeleton in Neurodevelopmental Disorders. Front. Cell. Neurosci. 2018, 12, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukata, M.; Watanabe, T.; Noritake, J.; Nakagawa, M.; Yamaga, M.; Kuroda, S.; Matsuura, Y.; Iwamatsu, A.; Perez, F.; Kaibuchi, K. Rac1 and Cdc42 capture microtubules through IQGAP1 and CLIP-170. Cell 2002, 109, 873–885. [Google Scholar] [CrossRef]
- Hedman, D.; Reza Barzegar, H.; Rosén, A.; Wågberg, T.; Andreas Larsson, J. On the Stability and Abundance of Single Walled Carbon Nanotubes. Sci. Rep. 2015, 5, 16850. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.-C.; Xiong, Z.-Q. Molecular and Synaptic Bases of CDKL5 Disorder. Dev Neurobiol 2019, 79, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, M.S.; Giarda, E.; Bedogni, F.; La Montanara, P.; Ricciardi, S.; Ciceri, D.; Alberio, T.; Landsberger, N.; Rusconi, L.; Kilstrup-Nielsen, C. CDKL5 and Shootin1 Interact and Concur in Regulating Neuronal Polarization. PLoS ONE 2016, 11, e0148634. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, C.; Medici, G.; Trazzi, S.; Gennaccaro, L.; Galvani, G.; Berteotti, C.; Ren, E.; Loi, M.; Ciani, E. CDKL5 deficiency predisposes neurons to cell death through the deregulation of SMAD3 signaling. Brain Pathol. 2019. [Google Scholar] [CrossRef]
- Conde, C.; Cáceres, A. Microtubule assembly, organization and dynamics in axons and dendrites. Nat. Rev. Neurosci. 2009, 10, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Nirschl, J.J.; Magiera, M.M.; Lazarus, J.E.; Janke, C.; Holzbaur, E.L.F. α-Tubulin Tyrosination and CLIP-170 Phosphorylation Regulate the Initiation of Dynein-Driven Transport in Neurons. Cell Rep. 2016, 14, 2637–2652. [Google Scholar] [CrossRef] [PubMed]
- Neukirchen, D.; Bradke, F. Cytoplasmic linker proteins regulate neuronal polarization through microtubule and growth cone dynamics. J. Neurosci. 2011, 31, 1528–1538. [Google Scholar] [CrossRef] [PubMed]
- Barbiero, I.; Peroni, D.; Siniscalchi, P.; Rusconi, L.; Tramarin, M.; De Rosa, R.; Motta, P.; Bianchi, M.; Kilstrup-Nielsen, C. Pregnenolone and pregnenolone-methyl-ether rescue neuronal defects caused by dysfunctional CLIP170 in a neuronal model of CDKL5 Deficiency Disorder. Neuropharmacology. under review.
- Lansbergen, G.; Komarova, Y.; Modesti, M.; Wyman, C.; Hoogenraad, C.C.; Goodson, H.V.; Lemaitre, R.P.; Drechsel, D.N.; van Munster, E.; Gadella, T.W.J.; et al. Conformational changes in CLIP-170 regulate its binding to microtubules and dynactin localization. J. Cell Biol. 2004, 166, 1003–1014. [Google Scholar] [CrossRef] [PubMed]
- Weng, J.-H.; Liang, M.-R.; Chen, C.-H.; Tong, S.-K.; Huang, T.-C.; Lee, S.-P.; Chen, Y.-R.; Chen, C.-T.; Chung, B.-C. Pregnenolone activates CLIP-170 to promote microtubule growth and cell migration. Nat. Chem. Biol. 2013, 9, 636–642. [Google Scholar] [CrossRef]
- Toriyama, M.; Shimada, T.; Kim, K.B.; Mitsuba, M.; Nomura, E.; Katsuta, K.; Sakumura, Y.; Roepstorff, P.; Inagaki, N. Shootin1: A protein involved in the organization of an asymmetric signal for neuronal polarization. J. Cell Biol. 2006, 175, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Kubo, Y.; Baba, K.; Toriyama, M.; Minegishi, T.; Sugiura, T.; Kozawa, S.; Ikeda, K.; Inagaki, N. Shootin1-cortactin interaction mediates signal-force transduction for axon outgrowth. J. Cell Biol. 2015, 210, 663–676. [Google Scholar] [CrossRef]
- Chen, Q.; Zhu, Y.-C.; Yu, J.; Miao, S.; Zheng, J.; Xu, L.; Zhou, Y.; Li, D.; Zhang, C.; Tao, J.; et al. CDKL5, a protein associated with rett syndrome, regulates neuronal morphogenesis via Rac1 signaling. J. Neurosci. 2010, 30, 12777–12786. [Google Scholar] [CrossRef] [PubMed]
- Amendola, E.; Zhan, Y.; Mattucci, C.; Castroflorio, E.; Calcagno, E.; Fuchs, C.; Lonetti, G.; Silingardi, D.; Vyssotski, A.L.; Farley, D.; et al. Mapping pathological phenotypes in a mouse model of CDKL5 disorder. PLoS ONE 2014, 9, e91613. [Google Scholar] [CrossRef]
- Tang, S.; Wang, I.-T.J.; Yue, C.; Takano, H.; Terzic, B.; Pance, K.; Lee, J.Y.; Cui, Y.; Coulter, D.A.; Zhou, Z. Loss of CDKL5 in Glutamatergic Neurons Disrupts Hippocampal Microcircuitry and Leads to Memory Impairment in Mice. J. Neurosci. 2017, 37, 7420–7437. [Google Scholar] [CrossRef] [Green Version]
- Okuda, K.; Takao, K.; Watanabe, A.; Miyakawa, T.; Mizuguchi, M.; Tanaka, T. Comprehensive behavioral analysis of the Cdkl5 knockout mice revealed significant enhancement in anxiety- and fear-related behaviors and impairment in both acquisition and long-term retention of spatial reference memory. PLoS ONE 2018, 13, e0196587. [Google Scholar] [CrossRef]
- Jacquemet, G.; Humphries, M.J. IQGAP1 is a key node within the small GTPase network. Small GTPases 2013, 4, 199–207. [Google Scholar] [CrossRef] [Green Version]
- Swiech, L.; Blazejczyk, M.; Urbanska, M.; Pietruszka, P.; Dortland, B.R.; Malik, A.R.; Wulf, P.S.; Hoogenraad, C.C.; Jaworski, J. CLIP-170 and IQGAP1 cooperatively regulate dendrite morphology. J. Neurosci. 2011, 31, 4555–4568. [Google Scholar] [CrossRef]
- Ramkumar, A.; Jong, B.Y.; Ori-McKenney, K.M. ReMAPping the microtubule landscape: How phosphorylation dictates the activities of microtubule-associated proteins. Dev. Dyn. 2018, 247, 138–155. [Google Scholar] [PubMed]
- Ricciardi, S.; Ungaro, F.; Hambrock, M.; Rademacher, N.; Stefanelli, G.; Brambilla, D.; Sessa, A.; Magagnotti, C.; Bachi, A.; Giarda, E.; et al. CDKL5 ensures excitatory synapse stability by reinforcing NGL-1-PSD95 interaction in the postsynaptic compartment and is impaired in patient iPSC-derived neurons. Nat. Cell Biol. 2012, 14, 911–923. [Google Scholar] [CrossRef] [PubMed]
- Della Sala, G.; Putignano, E.; Chelini, G.; Melani, R.; Calcagno, E.; Michele Ratto, G.; Amendola, E.; Gross, C.T.; Giustetto, M.; Pizzorusso, T. Dendritic Spine Instability in a Mouse Model of CDKL5 Disorder Is Rescued by Insulin-like Growth Factor 1. Biol. Psychiatry 2016, 80, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Lamprecht, R.; LeDoux, J. Structural plasticity and memory. Nat. Rev. Neurosci. 2004, 5, 45–54. [Google Scholar] [CrossRef] [PubMed]
- La Montanara, P.; Rusconi, L.; Locarno, A.; Forti, L.; Barbiero, I.; Tramarin, M.; Chandola, C.; Kilstrup-Nielsen, C.; Landsberger, N. Synaptic synthesis, dephosphorylation, and degradation: A novel paradigm for an activity-dependent neuronal control of CDKL5. J. Biol. Chem. 2015, 290, 4512–4527. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.-C.; Li, D.; Wang, L.; Lu, B.; Zheng, J.; Zhao, S.-L.; Zeng, R.; Xiong, Z.-Q. Palmitoylation-dependent CDKL5-PSD-95 interaction regulates synaptic targeting of CDKL5 and dendritic spine development. Proc. Natl. Acad. Sci. USA 2013, 110, 9118–9123. [Google Scholar] [CrossRef] [PubMed]
- Pizzo, R.; Gurgone, A.; Castroflorio, E.; Amendola, E.; Gross, C.; Sassoè-Pognetto, M.; Giustetto, M. Lack of Cdkl5 Disrupts the Organization of Excitatory and Inhibitory Synapses and Parvalbumin Interneurons in the Primary Visual Cortex. Front. Cell. Neurosci. 2016, 10, 261. [Google Scholar] [CrossRef]
- Tramarin, M.; Rusconi, L.; Pizzamiglio, L.; Barbiero, I.; Peroni, D.; Scaramuzza, L.; Guilliams, T.; Cavalla, D.; Antonucci, F.; Kilstrup-Nielsen, C. The antidepressant tianeptine reverts synaptic AMPA receptor defects caused by deficiency of CDKL5. Hum. Mol. Genet. 2018, 27, 2052–2063. [Google Scholar] [CrossRef] [PubMed]
- Ren, E.; Roncacé, V.; Trazzi, S.; Fuchs, C.; Medici, G.; Gennaccaro, L.; Loi, M.; Galvani, G.; Ye, K.; Rimondini, R.; et al. Functional and Structural Impairments in the Perirhinal Cortex of a Mouse Model of CDKL5 Deficiency Disorder Are Rescued by a TrkB Agonist. Front. Cell. Neurosci. 2019, 13, 169. [Google Scholar] [CrossRef] [PubMed]
- Yennawar, M.; White, R.S.; Jensen, F.E. AMPA Receptor Dysregulation and Therapeutic Interventions in a Mouse Model of CDKL5 Deficiency Disorder. J. Neurosci. 2019, 39, 4814–4828. [Google Scholar] [CrossRef] [Green Version]
- Ethell, I.M.; Pasquale, E.B. Molecular mechanisms of dendritic spine development and remodeling. Prog. Neurobiol. 2005, 75, 161–205. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Viesselmann, C.; Nam, S.; Merriam, E.; Dent, E.W. Activity-dependent dynamic microtubule invasion of dendritic spines. J. Neurosci. 2008, 28, 13094–13105. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Firestein, B.L.; Zheng, J.Q. Microtubules in dendritic spine development. J. Neurosci. 2008, 28, 12120–12124. [Google Scholar] [CrossRef] [PubMed]
- Schätzle, P.; Esteves da Silva, M.; Tas, R.P.; Katrukha, E.A.; Hu, H.Y.; Wierenga, C.J.; Kapitein, L.C.; Hoogenraad, C.C. Activity-Dependent Actin Remodeling at the Base of Dendritic Spines Promotes Microtubule Entry. Curr. Biol. 2018, 28, 2081–2093. [Google Scholar] [CrossRef] [PubMed]
- Pchitskaya, E.; Kraskovskaya, N.; Chernyuk, D.; Popugaeva, E.; Zhang, H.; Vlasova, O.; Bezprozvanny, I. Stim2-Eb3 Association and Morphology of Dendritic Spines in Hippocampal Neurons. Sci. Rep. 2017, 7, 17625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrick, C.; Fischer, A.; Srivastava, D.P.; Tronson, N.C.; Penzes, P.; Radulovic, J. N-cadherin regulates cytoskeletally associated IQGAP1/ERK signaling and memory formation. Neuron 2007, 55, 786–798. [Google Scholar] [CrossRef] [PubMed]
- Jausoro, I.; Mestres, I.; Quassollo, G.; Masseroni, L.; Heredia, F.; Caceres, A. Regulation of spine density and morphology by IQGAP1 protein domains. PLoS ONE 2013, 8, e56574. [Google Scholar] [CrossRef]
- Gao, C.; Frausto, S.F.; Guedea, A.L.; Tronson, N.C.; Jovasevic, V.; Leaderbrand, K.; Corcoran, K.A.; Guzmán, Y.F.; Swanson, G.T.; Radulovic, J. IQGAP1 regulates NR2A signaling, spine density, and cognitive processes. J. Neurosci. 2011, 31, 8533–8542. [Google Scholar] [CrossRef]
- Nuriya, M.; Oh, S.; Huganir, R.L. Phosphorylation-dependent interactions of alpha-Actinin-1/IQGAP1 with the AMPA receptor subunit GluR4. J. Neurochem. 2005, 95, 544–552. [Google Scholar] [CrossRef]
- Wang, S.-H.J.; Celic, I.; Choi, S.-Y.; Riccomagno, M.; Wang, Q.; Sun, L.O.; Mitchell, S.P.; Vasioukhin, V.; Huganir, R.L.; Kolodkin, A.L. Dlg5 regulates dendritic spine formation and synaptogenesis by controlling subcellular N-cadherin localization. J. Neurosci. 2014, 34, 12745–12761. [Google Scholar] [CrossRef]
- Sandí, M.-J.; Marshall, C.B.; Balan, M.; Coyaud, É.; Zhou, M.; Monson, D.M.; Ishiyama, N.; Chandrakumar, A.A.; La Rose, J.; Couzens, A.L.; et al. MARK3-mediated phosphorylation of ARHGEF2 couples microtubules to the actin cytoskeleton to establish cell polarity. Sci. Signal. 2017, 10, eaan3286. [Google Scholar] [CrossRef] [PubMed]
- McVicker, D.P.; Awe, A.M.; Richters, K.E.; Wilson, R.L.; Cowdrey, D.A.; Hu, X.; Chapman, E.R.; Dent, E.W. Transport of a kinesin-cargo pair along microtubules into dendritic spines undergoing synaptic plasticity. Nat. Commun. 2016, 7, 12741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schatten, H. The mammalian centrosome and its functional significance. Histochem. Cell Biol. 2008, 129, 667–686. [Google Scholar] [PubMed] [Green Version]
- Bettencourt-Dias, M.; Glover, D.M. Centrosome biogenesis and function: Centrosomics brings new understanding. Nat. Rev. Mol. Cell Biol. 2007, 8, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Nigg, E.A.; Raff, J.W. Centrioles, centrosomes, and cilia in health and disease. Cell 2009, 139, 663–678. [Google Scholar] [CrossRef]
- Glotzer, M. Cytokinesis: GAP gap. Curr. Biol. 2009, 19, R162–R165. [Google Scholar] [CrossRef]
- Lim, H.H.; Zhang, T.; Surana, U. Regulation of centrosome separation in yeast and vertebrates: Common threads. Trends Cell Biol. 2009, 19, 325–333. [Google Scholar] [CrossRef]
- Staples, C.J.; Myers, K.N.; Beveridge, R.D.D.; Patil, A.A.; Lee, A.J.X.; Swanton, C.; Howell, M.; Boulton, S.J.; Collis, S.J. The centriolar satellite protein Cep131 is important for genome stability. J. Cell. Sci. 2012, 125, 4770–4779. [Google Scholar] [CrossRef]
- Canning, P.; Park, K.; Gonçalves, J.; Li, C.; Howard, C.J.; Sharpe, T.D.; Holt, L.J.; Pelletier, L.; Bullock, A.N.; Leroux, M.R. CDKL Family Kinases Have Evolved Distinct Structural Features and Ciliary Function. Cell Rep. 2018, 22, 885–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graser, S.; Stierhof, Y.-D.; Lavoie, S.B.; Gassner, O.S.; Lamla, S.; Le Clech, M.; Nigg, E.A. Cep164, a novel centriole appendage protein required for primary cilium formation. J. Cell Biol. 2007, 179, 321–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malicki, J.J.; Johnson, C.A. The Cilium: Cellular Antenna and Central Processing Unit. Trends Cell Biol. 2017, 27, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Valente, E.M.; Rosti, R.O.; Gibbs, E.; Gleeson, J.G. Primary cilia in neurodevelopmental disorders. Nat. Rev. Neurol. 2014, 10, 27–36. [Google Scholar] [CrossRef]
- Tam, L.-W.; Ranum, P.T.; Lefebvre, P.A. CDKL5 regulates flagellar length and localizes to the base of the flagella in Chlamydomonas. Mol. Biol. Cell. 2013, 24, 588–600. [Google Scholar] [CrossRef] [PubMed]
- Hector, R.D.; Kalscheuer, V.M.; Hennig, F.; Leonard, H.; Downs, J.; Clarke, A.; Benke, T.A.; Armstrong, J.; Pineda, M.; Bailey, M.E.S.; et al. CDKL5 variants: Improving our understanding of a rare neurologic disorder. Neurol. Genet. 2017, 3, e200. [Google Scholar] [CrossRef] [PubMed]
- Bertani, I.; Rusconi, L.; Bolognese, F.; Forlani, G.; Conca, B.; De Monte, L.; Badaracco, G.; Landsberger, N.; Kilstrup-Nielsen, C. Functional consequences of mutations in CDKL5, an X-linked gene involved in infantile spasms and mental retardation. J. Biol. Chem. 2006, 281, 32048–32056. [Google Scholar] [CrossRef]
- Rusconi, L.; Salvatoni, L.; Giudici, L.; Bertani, I.; Kilstrup-Nielsen, C.; Broccoli, V.; Landsberger, N. CDKL5 expression is modulated during neuronal development and its subcellular distribution is tightly regulated by the C-terminal tail. J. Biol. Chem. 2008, 283, 30101–30111. [Google Scholar] [CrossRef] [PubMed]
- Rosas-Vargas, H.; Bahi-Buisson, N.; Philippe, C.; Nectoux, J.; Girard, B.; N’Guyen Morel, M.A.; Gitiaux, C.; Lazaro, L.; Odent, S.; Jonveaux, P.; et al. Impairment of CDKL5 nuclear localisation as a cause for severe infantile encephalopathy. J. Med. Genet. 2008, 45, 172–178. [Google Scholar] [CrossRef]
- Nectoux, J.; Heron, D.; Tallot, M.; Chelly, J.; Bienvenu, T. Maternal origin of a novel C-terminal truncation mutation in CDKL5 causing a severe atypical form of Rett syndrome. Clin. Genet. 2006, 70, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Russo, S.; Marchi, M.; Cogliati, F.; Bonati, M.T.; Pintaudi, M.; Veneselli, E.; Saletti, V.; Balestrini, M.; Ben-Zeev, B.; Larizza, L. Novel mutations in the CDKL5 gene, predicted effects and associated phenotypes. Neurogenetics 2009, 10, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.-T.J.; Allen, M.; Goffin, D.; Zhu, X.; Fairless, A.H.; Brodkin, E.S.; Siegel, S.J.; Marsh, E.D.; Blendy, J.A.; Zhou, Z. Loss of CDKL5 disrupts kinome profile and event-related potentials leading to autistic-like phenotypes in mice. Proc. Natl. Acad. Sci. USA 2012, 109, 21516–21521. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.; Terzic, B.; Wang, I.-T.J.; Sarmiento, N.; Sizov, K.; Cui, Y.; Takano, H.; Marsh, E.D.; Zhou, Z.; Coulter, D.A. Altered NMDAR signaling underlies autistic-like features in mouse models of CDKL5 deficiency disorder. Nat. Commun. 2019, 10, 2655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szafranski, P.; Golla, S.; Jin, W.; Fang, P.; Hixson, P.; Matalon, R.; Kinney, D.; Bock, H.-G.; Craigen, W.; Smith, J.L.; et al. Neurodevelopmental and neurobehavioral characteristics in males and females with CDKL5 duplications. Eur. J. Hum. Genet. 2015, 23, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Marchisella, F.; Coffey, E.T.; Hollos, P. Microtubule and microtubule associated protein anomalies in psychiatric disease. Cytoskeleton (Hoboken) 2016, 73, 596–611. [Google Scholar] [CrossRef] [PubMed]
- Brunden, K.R.; Lee, V.M.-Y.; Smith, A.B.; Trojanowski, J.Q.; Ballatore, C. Altered microtubule dynamics in neurodegenerative disease: Therapeutic potential of microtubule-stabilizing drugs. Neurobiol. Dis. 2017, 105, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Amos, L.A. Microtubule structure and its stabilisation. Org. Biomol. Chem. 2004, 2, 2153–2160. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Maiti, A.; Shively, S.; Lakhani, F.; McDonald-Jones, G.; Bruce, J.; Lee, E.B.; Xie, S.X.; Joyce, S.; Li, C.; et al. Microtubule-binding drugs offset tau sequestration by stabilizing microtubules and reversing fast axonal transport deficits in a tauopathy model. Proc. Natl. Acad. Sci. USA 2005, 102, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Brunden, K.R.; Ballatore, C.; Lee, V.M.-Y.; Smith, A.B.; Trojanowski, J.Q. Brain-penetrant microtubule-stabilizing compounds as potential therapeutic agents for tauopathies. Biochem. Soc. Trans. 2012, 40, 661–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Carroll, J.; Trojanowski, J.Q.; Yao, Y.; Iba, M.; Potuzak, J.S.; Hogan, A.-M.L.; Xie, S.X.; Ballatore, C.; Smith, A.B.; et al. The microtubule-stabilizing agent, epothilone D, reduces axonal dysfunction, neurotoxicity, cognitive deficits, and Alzheimer-like pathology in an interventional study with aged tau transgenic mice. J. Neurosci. 2012, 32, 3601–3611. [Google Scholar] [CrossRef] [PubMed]
- Brunden, K.R.; Zhang, B.; Carroll, J.; Yao, Y.; Potuzak, J.S.; Hogan, A.-M.L.; Iba, M.; James, M.J.; Xie, S.X.; Ballatore, C.; et al. Epothilone D improves microtubule density, axonal integrity, and cognition in a transgenic mouse model of tauopathy. J. Neurosci. 2010, 30, 13861–13866. [Google Scholar] [CrossRef] [PubMed]
- Gozes, I. NAP (davunetide) provides functional and structural neuroprotection. Curr. Pharm. Des. 2011, 17, 1040–1044. [Google Scholar] [CrossRef] [PubMed]
- Helsmoortel, C.; Vulto-van Silfhout, A.T.; Coe, B.P.; Vandeweyer, G.; Rooms, L.; van den Ende, J.; Schuurs-Hoeijmakers, J.H.M.; Marcelis, C.L.; Willemsen, M.H.; Vissers, L.E.L.M.; et al. A SWI/SNF-related autism syndrome caused by de novo mutations in ADNP. Nat. Genet. 2014, 46, 380–384. [Google Scholar] [CrossRef] [Green Version]
- Oz, S.; Kapitansky, O.; Ivashco-Pachima, Y.; Malishkevich, A.; Giladi, E.; Skalka, N.; Rosin-Arbesfeld, R.; Mittelman, L.; Segev, O.; Hirsch, J.A.; et al. The NAP motif of activity-dependent neuroprotective protein (ADNP) regulates dendritic spines through microtubule end binding proteins. Mol. Psychiatry 2014, 19, 1115–1124. [Google Scholar] [CrossRef] [Green Version]
- Gozes, I. ADNP Regulates Cognition: A Multitasking Protein. Front. Neurosci. 2018, 12, 873. [Google Scholar] [CrossRef] [Green Version]
- Oz, S.; Ivashko-Pachima, Y.; Gozes, I. The ADNP derived peptide, NAP modulates the tubulin pool: Implication for neurotrophic and neuroprotective activities. PLoS ONE 2012, 7, e51458. [Google Scholar]
- Gozes, I. The cytoskeleton as a drug target for neuroprotection: The case of the autism- mutated ADNP. Biol. Chem. 2016, 397, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.alzforum.org/ (accessed on 21 July 2019).
- Javitt, D.C.; Buchanan, R.W.; Keefe, R.S.E.; Kern, R.; McMahon, R.P.; Green, M.F.; Lieberman, J.; Goff, D.C.; Csernansky, J.G.; McEvoy, J.P.; et al. Effect of the neuroprotective peptide davunetide (AL-108) on cognition and functional capacity in schizophrenia. Schizophr. Res. 2012, 136, 25–31. [Google Scholar] [CrossRef]
- Weng, J.-H.; Chung, B.-C. Nongenomic actions of neurosteroid pregnenolone and its metabolites. Steroids 2016, 111, 54–59. [Google Scholar] [CrossRef]
- Flood, J.F.; Morley, J.E.; Roberts, E. Memory-enhancing effects in male mice of pregnenolone and steroids metabolically derived from it. Proc. Natl. Acad. Sci. USA 1992, 89, 1567–1571. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.S.; Park, J.; Marx, C.E.; Hynan, L.S.; Gardner, C.; Davila, D.; Nakamura, A.; Sunderajan, P.; Lo, A.; Holmes, T. A randomized, double-blind, placebo-controlled trial of pregnenolone for bipolar depression. Neuropsychopharmacology 2014, 39, 2867–2873. [Google Scholar] [CrossRef]
- Osuji, I.J.; Vera-Bolaños, E.; Carmody, T.J.; Brown, E.S. Pregnenolone for cognition and mood in dual diagnosis patients. Psychiatry Res. 2010, 178, 309–312. [Google Scholar] [CrossRef]
- Bianchi, M.; Baulieu, E.-E. 3β-Methoxy-pregnenolone (MAP4343) as an innovative therapeutic approach for depressive disorders. Proc. Natl. Acad. Sci. USA 2012, 109, 1713–1718. [Google Scholar] [CrossRef] [PubMed]
- Parésys, L.; Hoffmann, K.; Froger, N.; Bianchi, M.; Villey, I.; Baulieu, E.-E.; Fuchs, E. Effects of the Synthetic Neurosteroid: 3β-Methoxypregnenolone (MAP4343) on Behavioral and Physiological Alterations Provoked by Chronic Psychosocial Stress in Tree Shrews. Int. J. Neuropsychopharmacol. 2016, 19, pyv119. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Microtubule dynamics and post-translational modifications. (A) Microtubules (MTs) are composed of α- and β-tubulin heterodimers that assemble to linear proto-filaments that associate laterally, forming the hollow MT cylinder. Assembly, which is nucleated from the γ-tubulin ring, occurs in a polarised fashion with the β-tubulin subunit oriented towards the growing plus-end. Newly-incorporated tubulin is bound to guanosine triphosphate (GTP) that gets rapidly hydrolysed upon polymerisation, generating guanosine diphosphate (GDP)-bound tubulin along the MT lattice. (B) MTs are dynamic structures undergoing cycles of growth (polymerisation) and shrinkage (depolymerisation). The switch from growth to shrinkage is termed catastrophe, whereas rescue indicates the switch from shrinkage to growth. (C) The α-tubulin subunit can be acetylated on lysine (K) 40, which is located on the inner side of the MT. The C-terminal tail of α-tubulin projects away from the MT lattice and undergoes various post-translational modifications. The extreme tyrosine residue (Y) can be enzymatically removed, generating detyrosinated α-tubulin, which normally correlates with long-lived MTs. Further removal of the penultimate glutamate (E) generates ∆2-tubulin.
Figure 1.
Microtubule dynamics and post-translational modifications. (A) Microtubules (MTs) are composed of α- and β-tubulin heterodimers that assemble to linear proto-filaments that associate laterally, forming the hollow MT cylinder. Assembly, which is nucleated from the γ-tubulin ring, occurs in a polarised fashion with the β-tubulin subunit oriented towards the growing plus-end. Newly-incorporated tubulin is bound to guanosine triphosphate (GTP) that gets rapidly hydrolysed upon polymerisation, generating guanosine diphosphate (GDP)-bound tubulin along the MT lattice. (B) MTs are dynamic structures undergoing cycles of growth (polymerisation) and shrinkage (depolymerisation). The switch from growth to shrinkage is termed catastrophe, whereas rescue indicates the switch from shrinkage to growth. (C) The α-tubulin subunit can be acetylated on lysine (K) 40, which is located on the inner side of the MT. The C-terminal tail of α-tubulin projects away from the MT lattice and undergoes various post-translational modifications. The extreme tyrosine residue (Y) can be enzymatically removed, generating detyrosinated α-tubulin, which normally correlates with long-lived MTs. Further removal of the penultimate glutamate (E) generates ∆2-tubulin.

Figure 2.
The effect of CDKL5 deficiency on microtubule dynamics in axonal growth cones. (A) In the axon, microtubules (MTs) are organised in a polar fashion with the plus-ends oriented distally. The central domain, which is contiguous with the axonal shaft, is rich in MTs that are bound by plus-end tracking proteins (+TIPS) including CLIP170. The peripheral domain is rich in actin that extends into the filopodia. The transition zone, lying in between these two domains, contains antiparallel actin arcs that form a barrier against the invasion of MTs into the peripheral domain. (B) In the absence of CDKL5, CLIP170 is in its closed conformation and is less associated with the MT plus-ends impacting their capacity to engorge into the actin rich area and leading to cessation of axonal outgrowth.
Figure 2.
The effect of CDKL5 deficiency on microtubule dynamics in axonal growth cones. (A) In the axon, microtubules (MTs) are organised in a polar fashion with the plus-ends oriented distally. The central domain, which is contiguous with the axonal shaft, is rich in MTs that are bound by plus-end tracking proteins (+TIPS) including CLIP170. The peripheral domain is rich in actin that extends into the filopodia. The transition zone, lying in between these two domains, contains antiparallel actin arcs that form a barrier against the invasion of MTs into the peripheral domain. (B) In the absence of CDKL5, CLIP170 is in its closed conformation and is less associated with the MT plus-ends impacting their capacity to engorge into the actin rich area and leading to cessation of axonal outgrowth.

Figure 3.
The effect of CDKL5 deficiency on dendritic microtubule dynamics. In dendrites, microtubules (MTs) have mixed orientations with the plus-ends oriented both proximally and distally. CDKL5 phosphorylates MAP1S inhibiting its binding to MTs (lower left). In the absence of CDKL5 (lower right), the binding of MAP1S to MTs is accompanied by increased life-time of EB3 binding to the plus-ends.
Figure 3.
The effect of CDKL5 deficiency on dendritic microtubule dynamics. In dendrites, microtubules (MTs) have mixed orientations with the plus-ends oriented both proximally and distally. CDKL5 phosphorylates MAP1S inhibiting its binding to MTs (lower left). In the absence of CDKL5 (lower right), the binding of MAP1S to MTs is accompanied by increased life-time of EB3 binding to the plus-ends.

Figure 4.
The effect of CDKL5 deficiency on dendritic spine morphology. (A) Dendritic spines, which are enriched in actin, are invaded by microtubules (MTs) upon neuronal activation. The plus-end tracking proteins (+TIP), bound to the MT plus-ends, interact with actin-binding proteins facilitating the invasion. (B) In the absence of CDKL5, MTs are less dynamic and fail to invade the spines, impacting the activity-dependent structural changes that lead to the formation of mushroom-shaped spines.
Figure 4.
The effect of CDKL5 deficiency on dendritic spine morphology. (A) Dendritic spines, which are enriched in actin, are invaded by microtubules (MTs) upon neuronal activation. The plus-end tracking proteins (+TIP), bound to the MT plus-ends, interact with actin-binding proteins facilitating the invasion. (B) In the absence of CDKL5, MTs are less dynamic and fail to invade the spines, impacting the activity-dependent structural changes that lead to the formation of mushroom-shaped spines.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Barbiero, I.; De Rosa, R.; Kilstrup-Nielsen, C. Microtubules: A Key to Understand and Correct Neuronal Defects in CDKL5 Deficiency Disorder? Int. J. Mol. Sci. 2019, 20, 4075. https://doi.org/10.3390/ijms20174075
AMA Style
Barbiero I, De Rosa R, Kilstrup-Nielsen C. Microtubules: A Key to Understand and Correct Neuronal Defects in CDKL5 Deficiency Disorder? International Journal of Molecular Sciences. 2019; 20(17):4075. https://doi.org/10.3390/ijms20174075
Chicago/Turabian StyleBarbiero, Isabella, Roberta De Rosa, and Charlotte Kilstrup-Nielsen. 2019. "Microtubules: A Key to Understand and Correct Neuronal Defects in CDKL5 Deficiency Disorder?" International Journal of Molecular Sciences 20, no. 17: 4075. https://doi.org/10.3390/ijms20174075
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.