Human Liver-Derived Extracellular Matrix for the Culture of Distinct Human Primary Liver Cells

 , , , , , and
, , , , , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Sourcing of Fresh Human Liver Tissue
2.2. Isolation, Cryopreservation and Purification of Human Primary Liver Cells
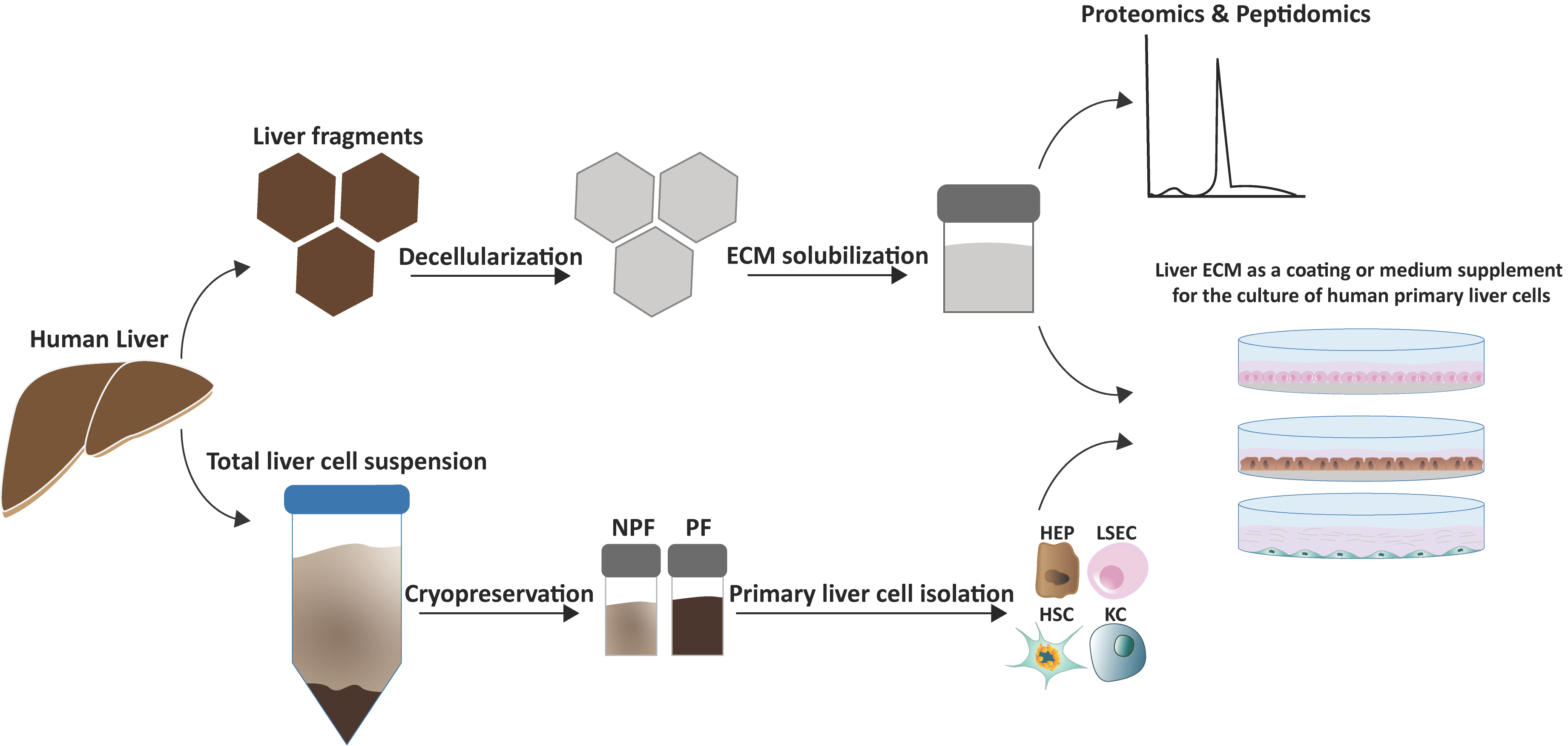
2.3. Human Liver Tissue Decellularization and Preparation of Solubilized HL-ECM
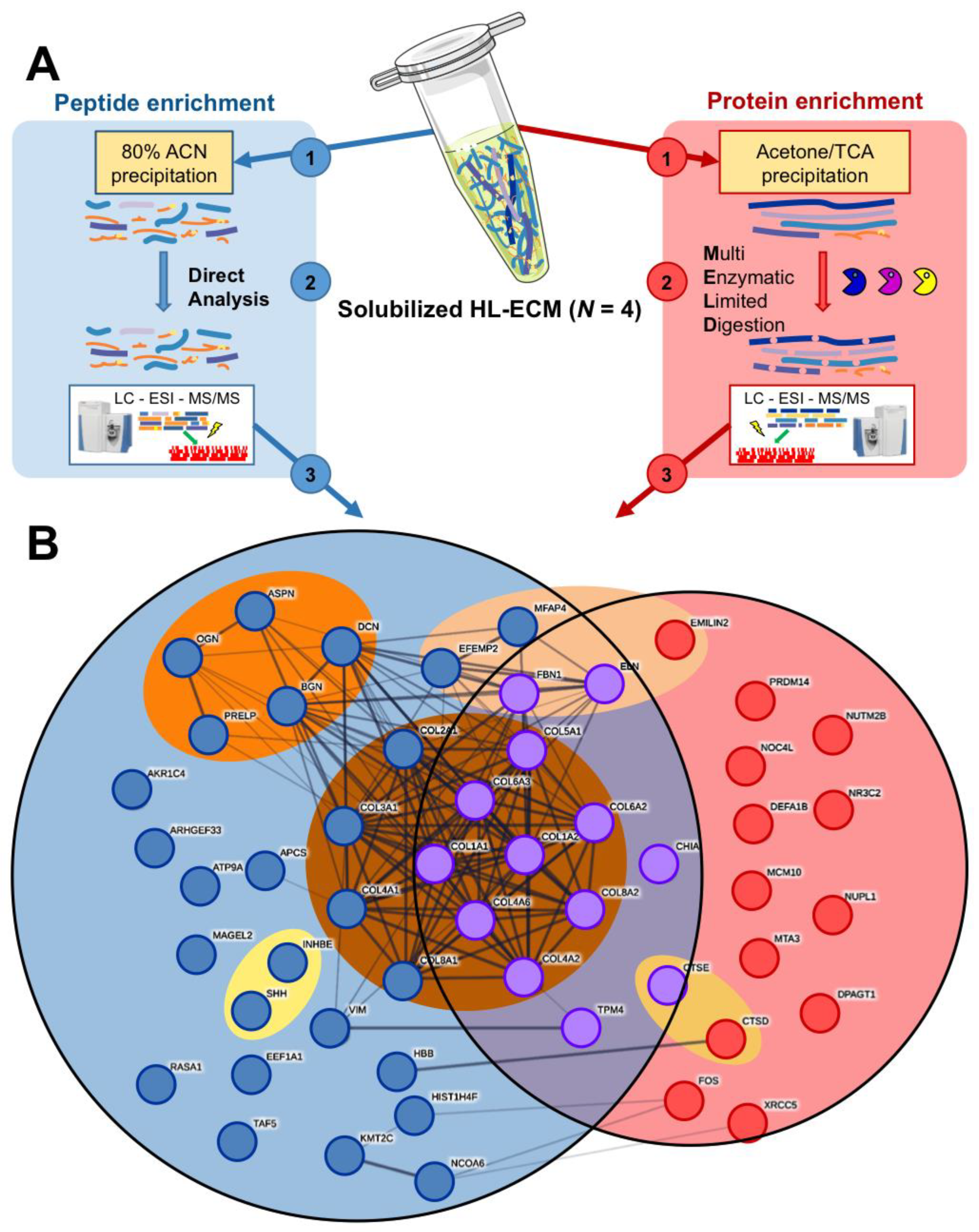
2.4. Proteomic Analysis of Lyophilized HL-ECM and Solubilized HL-ECM
2.4.1. Peptide Fraction Analysis
2.4.2. Protein Fraction Analysis
2.5. Electrophoresis
2.6. Functional Assays
2.7. RNA Extraction, Reverse Transcription and RT-qPCR
2.8. Statistical Analysis
3. Results
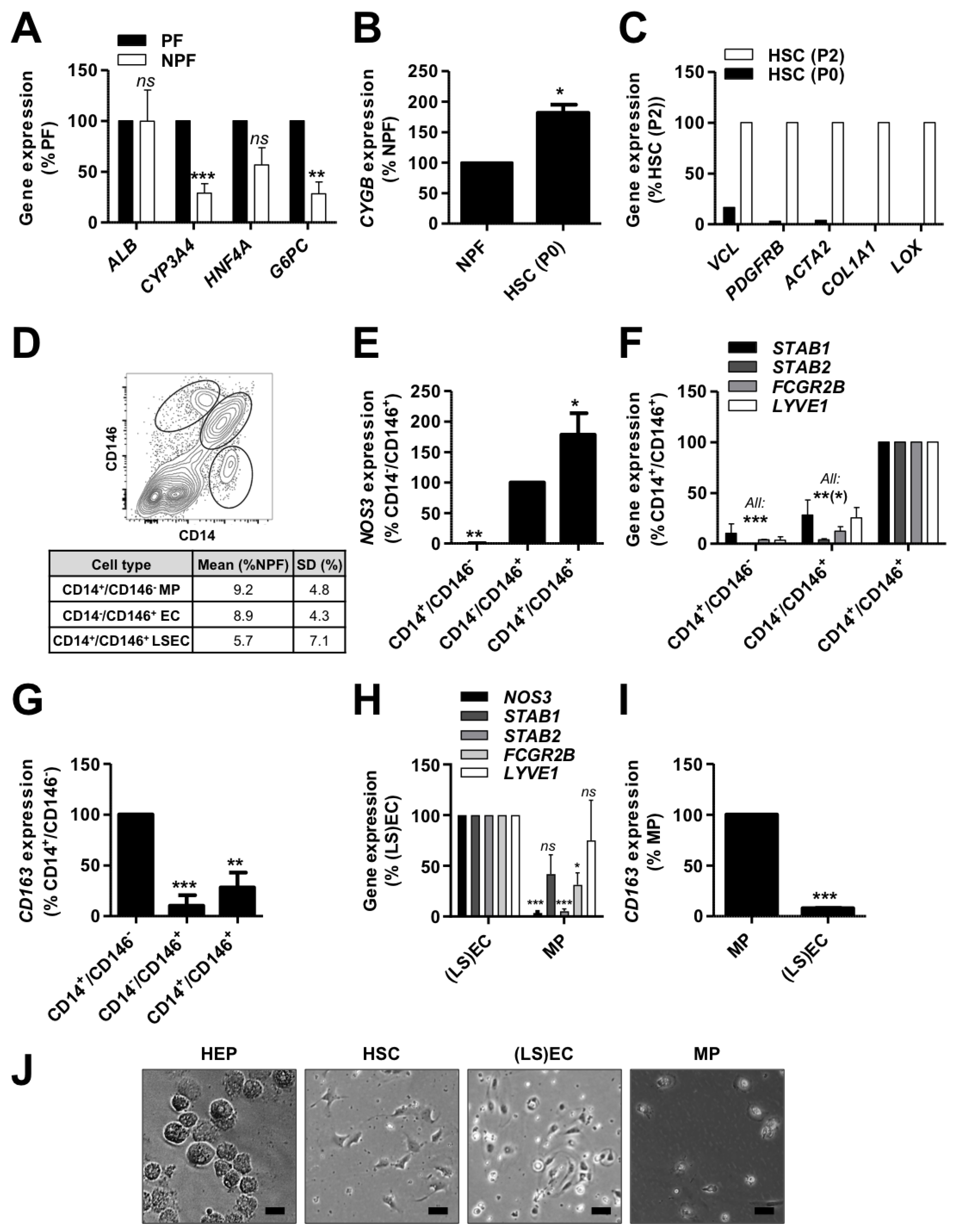
3.1. Enrichment of Human Liver Cells from Cryopreserved PF and NPF
3.2. Proteomic and Peptidomic Characterization of Solubilized HL-ECM
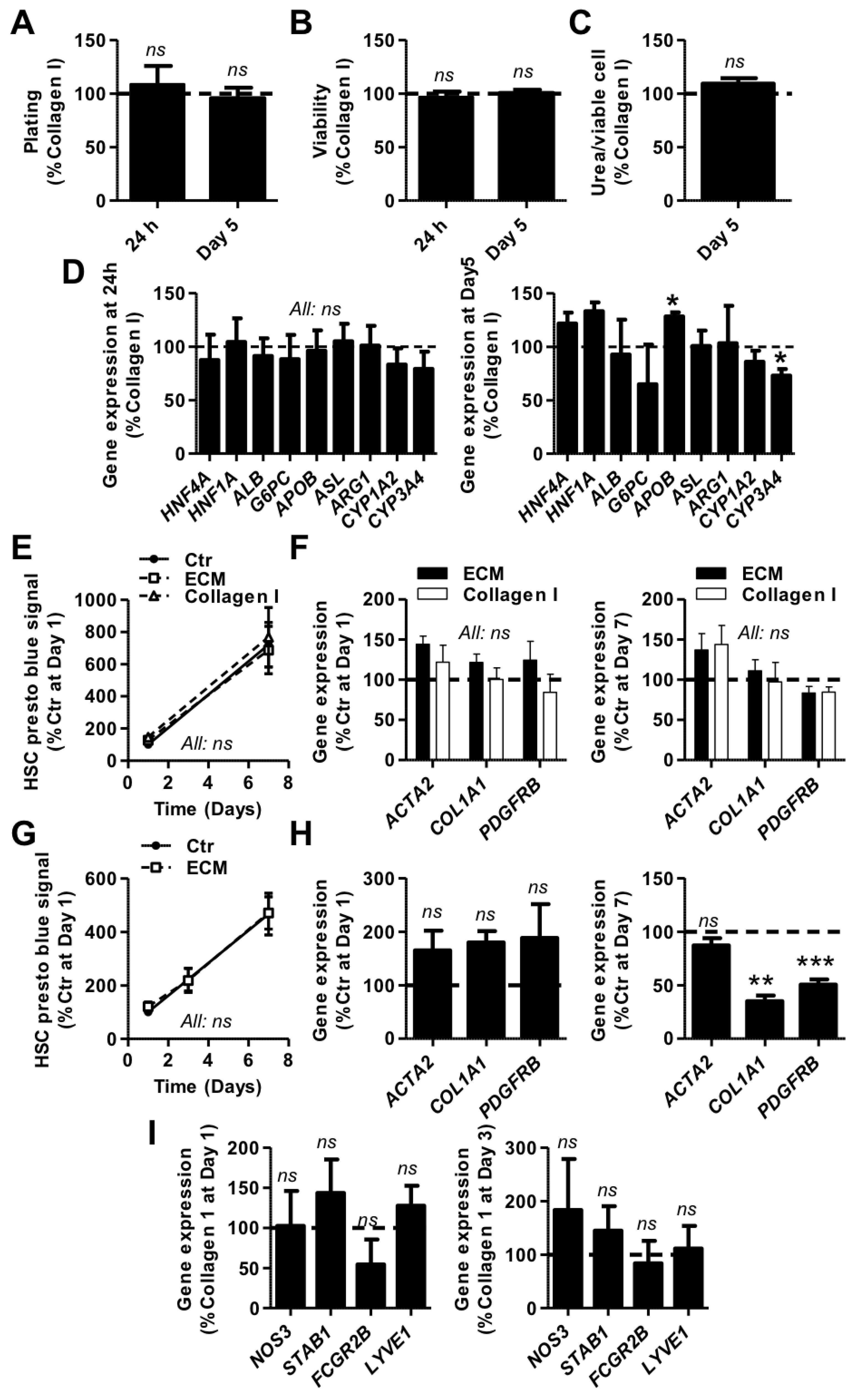
3.3. Evaluation of Solubilized HL-ECM for Primary Human Liver Cell Culture
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Soto-Gutierrez, A.; Navarro-Alvarez, N.; Kobayashi, N. Hepatocytes. In Molecular Pathology of Liver Diseases; Monga, S.P.S., Ed.; Springer: New York, NY, USA, 2011; pp. 17–26. [Google Scholar]
- Manco, R.; Leclercq, I.A.; Clerbaux, L.A. Liver Regeneration: Different Sub-Populations of Parenchymal Cells at Play Choreographed by an Injury-Specific Microenvironment. Int. J. Mol. Sci. 2018, 19, 4115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Wang, X.; Xie, G.; Wang, L.; Hill, C.K.; DeLeve, L.D. Liver sinusoidal endothelial cell progenitor cells promote liver regeneration in rats. J. Clin. Investig. 2012, 122, 1567–1573. [Google Scholar] [CrossRef] [PubMed]
- Ju, C.; Tacke, F. Hepatic macrophages in homeostasis and liver diseases: From pathogenesis to novel therapeutic strategies. Cell. Mol. Immunol. 2016, 13, 316–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuppan, D.; Kim, Y.O. Evolving therapies for liver fibrosis. J. Clin. Investig. 2013, 123, 1887–1901. [Google Scholar] [CrossRef] [Green Version]
- Stephenne, X.; Najimi, M.; Sokal, E.M. Hepatocyte cryopreservation: Is it time to change the strategy? World J. Gastroenterol. 2010, 16, 1–14. [Google Scholar]
- Zeigerer, A.; Wuttke, A.; Marsico, G.; Seifert, S.; Kalaidzidis, Y.; Zerial, M. Functional properties of hepatocytes in vitro are correlated with cell polarity maintenance. Exp. Cell Res. 2017, 350, 242–252. [Google Scholar] [CrossRef]
- Shulman, M.; Nahmias, Y. Long-term culture and coculture of primary rat and human hepatocytes. Methods Mol. Biol. 2013, 945, 287–302. [Google Scholar]
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef]
- El Taghdouini, A.; Najimi, M.; Sancho-Bru, P.; Sokal, E.; van Grunsven, L.A. In vitro reversion of activated primary human hepatic stellate cells. Fibrogenesis Tissue Repair 2015, 8, 14. [Google Scholar] [CrossRef] [Green Version]
- DeLeve, L.D.; Wang, X.; Hu, L.; McCuskey, M.K.; McCuskey, R.S. Rat liver sinusoidal endothelial cell phenotype is maintained by paracrine and autocrine regulation. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G757–G763. [Google Scholar] [CrossRef] [Green Version]
- March, S.; Hui, E.E.; Underhill, G.H.; Khetani, S.; Bhatia, S.N. Microenvironmental regulation of the sinusoidal endothelial cell phenotype in vitro. Hepatology 2009, 50, 920–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazza, G.; Rombouts, K.; Hall, A.R.; Urbani, L.; Luong, T.V.; Al-Akkad, W.; Longato, L.; Brown, D.; Maghsoudlou, P.; Dhillon, A.P.; et al. Decellularized human liver as a natural 3D-scaffold for liver bioengineering and transplantation. Sci. Rep. 2015, 5, 13079. [Google Scholar] [CrossRef] [PubMed]
- Sellaro, T.L.; Ranade, A.; Faulk, D.M.; McCabe, G.P.; Dorko, K.; Badylak, S.F.; Strom, S.C. Maintenance of human hepatocyte function in vitro by liver-derived extracellular matrix gels. Tissue Eng. Part A 2010, 16, 1075–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sellaro, T.L.; Ravindra, A.K.; Stolz, D.B.; Badylak, S.F. Maintenance of hepatic sinusoidal endothelial cell phenotype in vitro using organ-specific extracellular matrix scaffolds. Tissue Eng. 2007, 13, 2301–2310. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Shin, J.; Park, H.M.; Kim, Y.G.; Kim, B.G.; Oh, J.W.; Cho, S.W. Liver extracellular matrix providing dual functions of two-dimensional substrate coating and three-dimensional injectable hydrogel platform for liver tissue engineering. Biomacromolecules 2014, 15, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Loneker, A.E.; Faulk, D.M.; Hussey, G.S.; D’Amore, A.; Badylak, S.F. Solubilized liver extracellular matrix maintains primary rat hepatocyte phenotype in-vitro. J. Biomed. Mater. Res. A 2016, 104, 957–965. [Google Scholar] [CrossRef]
- Coronado, R.E.; Somaraki-Cormier, M.; Natesan, S.; Christy, R.J.; Ong, J.L.; Halff, G.A. Decellularization and Solubilization of Porcine Liver for Use as a Substrate for Porcine Hepatocyte Culture: Method Optimization and Comparison. Cell Transpl. 2017, 26, 1840–1854. [Google Scholar] [CrossRef] [Green Version]
- Najimi, M.; Khuu, D.N.; Lysy, P.A.; Jazouli, N.; Abarca, J.; Sempoux, C.; Sokal, E.M. Adult-derived human liver mesenchymal-like cells as a potential progenitor reservoir of hepatocytes? Cell Transpl. 2007, 16, 717–728. [Google Scholar] [CrossRef]
- Coppin, L.; Sokal, E.; Stephenne, X. Hepatocyte Transplantation in Children. Methods Mol. Biol. 2017, 1506, 295–315. [Google Scholar]
- Wiederkehr, J.C.; Igreja, M.R.; Nogara, M.S.; Goncalves, N.; Montemezzo, G.P.; Wiederkehr, H.A.; Wassen, M.P.; Nobrega, H.A.; Zenatti, K.B.; Mori, L.Y.; et al. Use of IGL-1 preservation solution in liver transplantation. Transpl. Proc. 2014, 46, 1809–1811. [Google Scholar] [CrossRef]
- Werner, M.; Driftmann, S.; Kleinehr, K.; Kaiser, G.M.; Mathe, Z.; Treckmann, J.W.; Paul, A.; Skibbe, K.; Timm, J.; Canbay, A.; et al. All-In-One: Advanced preparation of Human Parenchymal and Non-Parenchymal Liver Cells. PLoS ONE 2015, 10, e0138655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crapo, P.M.; Gilbert, T.W.; Badylak, S.F. An overview of tissue and whole organ decellularization processes. Biomaterials 2011, 32, 3233–3243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viswanath, A.; Vanacker, J.; Germain, L.; Leprince, J.G.; Diogenes, A.; Shakesheff, K.M.; White, L.J.; Rieux, A.D. Extracellular matrix-derived hydrogels for dental stem cell delivery. J. Biomed. Mater. Res. A 2017, 105, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Morsa, D.; Baiwir, D.; la Rocca, R.; Zimmerman, T.A.; Hanozin, E.; Grifnee, E.; Longuespee, R.; Meuwis, M.A.; Smargiasso, N.; Pauw, E.; et al. Multi-Enzymatic Limited Digestion: The Next-Generation Sequencing for Proteomics? J. Proteome Res. 2019, 18, 2501–2513. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.C.; Gorenstein, M.V.; Li, G.Z.; Vissers, J.P.; Geromanos, S.J. Absolute quantification of proteins by LCMSE: A virtue of parallel MS acquisition. Mol. Cell. Proteom. 2006, 5, 144–156. [Google Scholar] [CrossRef] [Green Version]
- Saheli, M.; Sepantafar, M.; Pournasr, B.; Farzaneh, Z.; Vosough, M.; Piryaei, A.; Baharvand, H. Three-dimensional liver-derived extracellular matrix hydrogel promotes liver organoids function. J. Cell. Biochem. 2018, 119, 4320–4333. [Google Scholar] [CrossRef]
- Persico, M.; Masarone, M.; Damato, A.; Ambrosio, M.; Federico, A.; Rosato, V.; Bucci, T.; Carrizzo, A.; Vecchione, C. Nonalcoholic fatty liver disease and eNOS dysfunction in humans. BMC Gastroenterol. 2017, 17, 35. [Google Scholar]
- Motoyama, H.; Komiya, T.; Thuy le, T.T.; Tamori, A.; Enomoto, M.; Morikawa, H.; Iwai, S.; Uchida-Kobayashi, S.; Fujii, H.; Hagihara, A.; et al. Cytoglobin is expressed in hepatic stellate cells, but not in myofibroblasts, in normal and fibrotic human liver. Lab. Investig. 2014, 94, 192–207. [Google Scholar] [CrossRef] [Green Version]
- Strauss, O.; Phillips, A.; Ruggiero, K.; Bartlett, A.; Dunbar, P.R. Immunofluorescence identifies distinct subsets of endothelial cells in the human liver. Sci. Rep. 2017, 7, 44356. [Google Scholar] [CrossRef] [Green Version]
- Riccalton-Banks, L.; Bhandari, R.; Fry, J.; Shakesheff, K.M. A simple method for the simultaneous isolation of stellate cells and hepatocytes from rat liver tissue. Mol. Cell. Biochem. 2003, 248, 97–102. [Google Scholar] [CrossRef]
- Mederacke, I.; Dapito, D.H.; Affò, S.; Uchinami, H.; Schwabe, R.F. High-yield and high-purity isolation of hepatic stellate cells from normal and fibrotic mouse livers. Nat. Protoc. 2015, 10, 305–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilpin, A.; Yang, Y. Decellularization Strategies for Regenerative Medicine: From Processing Techniques to Applications. Biomed. Res. Int. 2017, 2017, 9831534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saldin, L.T.; Cramer, M.C.; Velankar, S.S.; White, L.J.; Badylak, S.F. Extracellular matrix hydrogels from decellularized tissues: Structure and function. Acta Biomater. 2017, 49, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naba, A.; Clauser, K.R.; Ding, H.; Whittaker, C.A.; Carr, S.A.; Hynes, R.O. The extracellular matrix: Tools and insights for the “omics” era. Matrix Biol. 2016, 49, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Naba, A.; Clauser, K.R.; Whittaker, C.A.; Carr, S.A.; Tanabe, K.K.; Hynes, R.O. Extracellular matrix signatures of human primary metastatic colon cancers and their metastases to liver. BMC Cancer 2014, 14, 518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wells, R.G. The role of matrix stiffness in hepatic stellate cell activation and liver fibrosis. J. Clin. Gastroenterol. 2005, 39, S158–S161. [Google Scholar] [CrossRef]
- Rohn, F.; Kordes, C.; Castoldi, M.; Gotze, S.; Poschmann, G.; Stuhler, K.; Herebian, D.; Benk, A.S.; Geiger, F.; Zhang, T.; et al. Laminin-521 promotes quiescence in isolated stellate cells from rat liver. Biomaterials 2018, 180, 36–51. [Google Scholar] [CrossRef]
- Stone, L.C.; Thorne, L.S.; Weston, C.J.; Graham, M.; Hodges, N.J. Cytoglobin expression in the hepatic stellate cell line HSC-T6 is regulated by extracellular matrix proteins dependent on FAK-signalling. Fibrogenesis Tissue Repair 2015, 8, 15. [Google Scholar] [CrossRef] [Green Version]
- Poisson, J.; Lemoinne, S.; Boulanger, C.; Durand, F.; Moreau, R.; Valla, D.; Rautou, P.E. Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J. Hepatol. 2017, 66, 212–227. [Google Scholar] [CrossRef] [Green Version]
- Dong, W.; Lu, A.; Zhao, J.; Yin, S.; Ou, B.; Feng, H. An efficient and simple co-culture method for isolating primary human hepatic cells: Potential application for tumor microenvironment research. Oncol. Rep. 2016, 36, 2126–2134. [Google Scholar] [CrossRef]
- Pfeiffer, E.; Kegel, V.; Zeilinger, K.; Hengstler, J.G.; Nussler, A.K.; Seehofer, D.; Damm, G. Featured Article: Isolation, characterization, and cultivation of human hepatocytes and non-parenchymal liver cells. Exp. Biol. Med. 2015, 240, 645–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, C.C.; Hendriks, D.F.; Moro, S.M.; Ellis, E.; Walsh, J.; Renblom, A.; Puigvert, L.F.; Dankers, A.C.; Jacobs, F.; Snoeys, J.; et al. Characterization of primary human hepatocyte spheroids as a model system for drug-induced liver injury, liver function and disease. Sci. Rep. 2016, 6, 25187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domansky, K.; Inman, W.; Serdy, J.; Dash, A.; Lim, M.H.; Griffith, L.G. Perfused multiwell plate for 3D liver tissue engineering. Lab Chip 2010, 10, 51–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattei, G.; Magliaro, C.; Pirone, A.; Ahluwalia, A. Decellularized Human Liver Is Too Heterogeneous for Designing a Generic Extracellular Matrix Mimic Hepatic Scaffold. Artif. Organs 2017, 41, E347–E355. [Google Scholar] [CrossRef] [PubMed]
- Kocabayoglu, P.; Lade, A.; Lee, Y.A.; Dragomir, A.C.; Sun, X.; Fiel, M.I.; Thung, S.; Aloman, C.; Soriano, P.; Hoshida, Y.; et al. beta-PDGF receptor expressed by hepatic stellate cells regulates fibrosis in murine liver injury, but not carcinogenesis. J. Hepatol. 2015, 63, 141–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinzani, M.; Marra, F. Cytokine receptors and signaling in hepatic stellate cells. Semin. Liver Dis. 2001, 21, 397–416. [Google Scholar] [CrossRef] [PubMed]
- Nagao, R.J.; Xu, J.; Luo, P.; Xue, J.; Wang, Y.; Kotha, S.; Zeng, W.; Fu, X.; Himmelfarb, J.; Zheng, Y. Decellularized Human Kidney Cortex Hydrogels Enhance Kidney Microvascular Endothelial Cell Maturation and Quiescence. Tissue Eng. Part A 2016, 22, 1140–1150. [Google Scholar] [CrossRef] [Green Version]
- Sackett, S.D.; Tremmel, D.M.; Ma, F.; Feeney, A.K.; Maguire, R.M.; Brown, M.E.; Zhou, Y.; Li, X.; O’Brien, C.; Li, L.; et al. Extracellular matrix scaffold and hydrogel derived from decellularized and delipidized human pancreas. Sci. Rep. 2018, 8, 10452. [Google Scholar] [CrossRef]
- Wang, J.K.; Luo, B.; Guneta, V.; Li, L.; Foo, S.E.M.; Dai, Y.; Tan, T.T.Y.; Tan, N.S.; Choong, C.; Wong, M.T.C. Supercritical carbon dioxide extracted extracellular matrix material from adipose tissue. Mater. Sci. Eng. C Mater. Biol. Appl. 2017, 75, 349–358. [Google Scholar] [CrossRef]
- Wehmeyer, J.L.; Natesan, S.; Christy, R.J. Development of a Sterile Amniotic Membrane Tissue Graft Using Supercritical Carbon Dioxide. Tissue Eng. Part C Methods 2015, 21, 649–659. [Google Scholar] [CrossRef]
- Chen, R.N.; Ho, H.O.; Tsai, Y.T.; Sheu, M.T. Process development of an acellular dermal matrix (ADM) for biomedical applications. Biomaterials 2004, 25, 2679–2686. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhang, J.; Wang, R.; Gu, Y.; Li, J.; Wang, C. Triton X-100 combines with chymotrypsin: A more promising protocol to prepare decellularized porcine carotid arteries. Biomed. Mater. Eng. 2017, 28, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Zhou, Y.; Li, N.; Huang, M.; Duan, H.; Ge, J.; Xiang, P.; Wang, Z. The use of phospholipase A(2) to prepare acellular porcine corneal stroma as a tissue engineering scaffold. Biomaterials 2009, 30, 3513–3522. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Protein | Division | Function | Fraction |
|---|---|---|---|---|
| COL1A1 | Collagen I α1 chain | Core matrisome | Collagen | Both |
| COL1A2 | Collagen I α2 chain | |||
| COL2A1 | Collagen II α1 chain | |||
| COL3A1 | Collagen III α1 chain | |||
| COL4A2 | Collagen IV α2 chain | |||
| COL4A6 | Collagen IV α6 chain | |||
| COL5A1 | Collagen V α1 chain | |||
| COL6A3 | Collagen VI α3 chain | |||
| COL4A1 | Collagen IV α1 chain | Peptide | ||
| COL6A2 | Collagen VI α2 chain | |||
| COL8A1 | Collagen VIII α1 chain | |||
| COL8A2 | Collagen VIII α2 chain | |||
| EMILIN2 | EMILIN 2 | Core matrisome | ECM glycoprotein | Protein |
| ELN | Elastin | Both | ||
| FBN1 | Fibrillin 1 | |||
| EFEMP2 | EGF-containing fibulin-like ECM protein | Peptide | ||
| MFAP4 | Microfibril-associated glycoprotein 4 | |||
| ASPN | Asporin | Core matrisome | Proteoglycan | Peptide |
| BGN | Biglycan | |||
| DCN | Decorin | |||
| OGN | Mimecan | |||
| PRELP | Prolargin | |||
| VTN | Vitronectin | |||
| CTSD | Cathepsin D | Matrisome-associated | ECM regulator | Protein |
| CTSE | Cathepsin E | Both | ||
| SHH | Sonic hedgehog | Matrisome-associated | Secreted factor | Peptide |
| INHBE | Inhibin β E | |||
| NR3C2 | Mineralocorticoid receptor | Matrisome-unrelated | Membrane receptor | Protein |
| DEFA1 | Neutrophil defensin 1 | Matrisome-unrelated | Secreted protein | Protein |
| CHIA | Acidic mammalian chitinase | Both | ||
| APCS | Serum amyloid P-component | Peptide | ||
| HBB | Hemoglobin subunit β | |||
| DPAGT1 | DPAG phosphotransferase | Intracellular | Protein | |
| FOS | Proto-oncogene c-Fos | |||
| MCM10 | MCM 10 | |||
| MTA3 | Metastasis-associated protein 3 | |||
| NOC4L | Nucleolar complex protein 4 | |||
| NUP58 | Nucleoporin p58/p45 | |||
| NUTM2B | NUT family member 2B | |||
| PRDM14 | PR domain zinc finger protein 14 | |||
| XRCC5 | X-ray repair cross-complementing protein 5 | |||
| TPM4 | Tropomyosin α4 chain | Both | ||
| ARHGEF33 | Rho guanine nucleotide exchange factor 33 | Peptide | ||
| ATP9A | Probable phospholipid-transporting ATPase IIA | |||
| EEF1A | Elongation factor 1α | |||
| KMT2C | Histone-lysine N-methyltransferase 2C | |||
| HIST1H4 | Histone H4 | |||
| MAGEL2 | MAGE-like protein 2 | |||
| NCOA6 | Nuclear receptor coactivator 6 | |||
| RASA1 | Ras GTPase-activating protein 1 | |||
| TAF5 | Transcription initiation factor TFIID subunit 5 | |||
| VIM | Vimentin | |||
| KRT1 | Keratin type II cytoskeletal 1 | Probable contaminant | Protein | |
| KRT9 | Keratin type I cytoskeletal 9 | |||
| KRT10 | Keratin type I cytoskeletal 10 | |||
| ALB (B. taurus) | Serum albumin | Contaminant | Both | |
| PGA4 | Pepsin A4 | |||
| DNASE1 | Deoxyribonuclease 1 | Peptide | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alevra Sarika, N.; Payen, V.L.; Fléron, M.; Ravau, J.; Brusa, D.; Najimi, M.; De Pauw, E.; Eppe, G.; Mazzucchelli, G.; Sokal, E.M.; et al. Human Liver-Derived Extracellular Matrix for the Culture of Distinct Human Primary Liver Cells. Cells 2020, 9, 1357. https://doi.org/10.3390/cells9061357
Alevra Sarika N, Payen VL, Fléron M, Ravau J, Brusa D, Najimi M, De Pauw E, Eppe G, Mazzucchelli G, Sokal EM, et al. Human Liver-Derived Extracellular Matrix for the Culture of Distinct Human Primary Liver Cells. Cells. 2020; 9(6):1357. https://doi.org/10.3390/cells9061357
Chicago/Turabian StyleAlevra Sarika, Niki, Valéry L. Payen, Maximilien Fléron, Joachim Ravau, Davide Brusa, Mustapha Najimi, Edwin De Pauw, Gauthier Eppe, Gabriel Mazzucchelli, Etienne M. Sokal, and et al. 2020. "Human Liver-Derived Extracellular Matrix for the Culture of Distinct Human Primary Liver Cells" Cells 9, no. 6: 1357. https://doi.org/10.3390/cells9061357