 Alexandra Njegic
Alexandra Njegic Claire Wilson
Claire Wilson Elizabeth J. Cartwright
Elizabeth J. Cartwright- 1Division of Cardiovascular Sciences, The University of Manchester, Manchester, United Kingdom
- 2Centre for Tumour Biology, Barts Cancer Institute, Queen Mary University of London, London, United Kingdom
- 3Institute of Translational Medicine, University of Liverpool, Liverpool, United Kingdom
Diseases of the heart, such as heart failure and cardiac arrhythmias, are a growing socio-economic burden. Calcium (Ca2+) dysregulation is key hallmark of the failing myocardium and has long been touted as a potential therapeutic target in the treatment of a variety of cardiovascular diseases (CVD). In the heart, Ca2+ is essential for maintaining normal cardiac function through the generation of the cardiac action potential and its involvement in excitation contraction coupling. As such, the proteins which regulate Ca2+ cycling and signaling play a vital role in maintaining Ca2+ homeostasis. Changes to the expression levels and function of Ca2+-channels, pumps and associated intracellular handling proteins contribute to altered Ca2+ homeostasis in CVD. The remodeling of Ca2+-handling proteins therefore results in impaired Ca2+ cycling, Ca2+ leak from the sarcoplasmic reticulum and reduced Ca2+ clearance, all of which contributes to increased intracellular Ca2+. Currently, approved treatments for targeting Ca2+ handling dysfunction in CVD are focused on Ca2+ channel blockers. However, whilst Ca2+ channel blockers have been successful in the treatment of some arrhythmic disorders, they are not universally prescribed to heart failure patients owing to their ability to depress cardiac function. Despite the progress in CVD treatments, there remains a clear need for novel therapeutic approaches which are able to reverse pathophysiology associated with heart failure and arrhythmias. Given that heart failure and cardiac arrhythmias are closely associated with altered Ca2+ homeostasis, this review will address the molecular changes to proteins associated with both Ca2+-handling and -signaling; their potential as novel therapeutic targets will be discussed in the context of pre-clinical and, where available, clinical data.
Introduction
Cardiovascular diseases, such as ischemic heart disease, stroke, and arrhythmias, are the leading cause of death globally (Roth et al., 2017); with over 17 million fatalities attributed annually to CVD (Collaborators GBDCoD., 2018). Acquired CVD are thought to arise due to a combination of modifiable environmental causes and underlying genetic pre-dispositions (Kannel et al., 1961; Khot et al., 2003). CVDs lead to both functional and structural changes to the vasculature and myocardium. In the heart, cardiac dysfunction encompassing cardiac decompensation, arrhythmogenesis and impaired contractility are all closely associated with changes to calcium (Ca2+) handling proteins, Ca2+ channels and Ca2+ pumps (Lou et al., 2012). Determining the full scope of changes to Ca2+-handling proteins and mechanisms will aid in the development of novel therapeutics to reverse and/or prevent CVD. This review will focus on how changes to Ca2+-handling proteins observed during cardiac arrhythmogenesis and HF have been therapeutically targeted, with emphasis placed on recent developments in the search for novel therapies.
Ca2+ and Ca2+-Handling Proteins in the Heart
Maintenance of Ca2+ Homeostasis
The initiation and control of each heartbeat is achieved by the cardiac conduction system consisting of the SAN, AVN, and His-Purkinje system. Electrical impulses, termed cardiac AP arise from the SAN, often referred to as the pacemaker of heart, and propagate throughout the heart (Boyett, 2009). During the cardiac AP, the electrical membrane potential rapidly rises and falls, referred to as depolarization and repolarization respectively, triggering ECC. The changes in membrane potential are driven by the movement of ions across the cell membrane (Grant, 2009). Generally, the cardiac AP is divided into five phases (Figure 1): (Phase 0) involves rapid depolarization of the membrane potential which is key to the propagation of cardiac impulse in the heart; (Phase 1) is a brief interval of rapid depolarization; (Phase 2) known as the plateau phase of the cardiac AP in which Ca2+ enters the cell to trigger ECC; (Phase 3) rapid repolarization restores the membrane potential; and (Phase 4) the membrane potential is stable at the resting potential (Grant, 2009). Following an AP, the cell enters a recovery state termed the refractory period. Here, the cell is unable to initiate an AP which allows the heart to prepare for the next contraction (Ramza et al., 1990). The key ionic currents involved in the generation and propagation of the cardiac AP are the Ca2+ (ICa), sodium (Na+, INa), and potassium (K+, IK) currents (Grant, 2009). It is worth noting that different cell types within the cardiac conduction system exhibit specific cardiac AP relating to their function. Differences in ion channel expression results in regional specific APs (Monfredi et al., 2010).
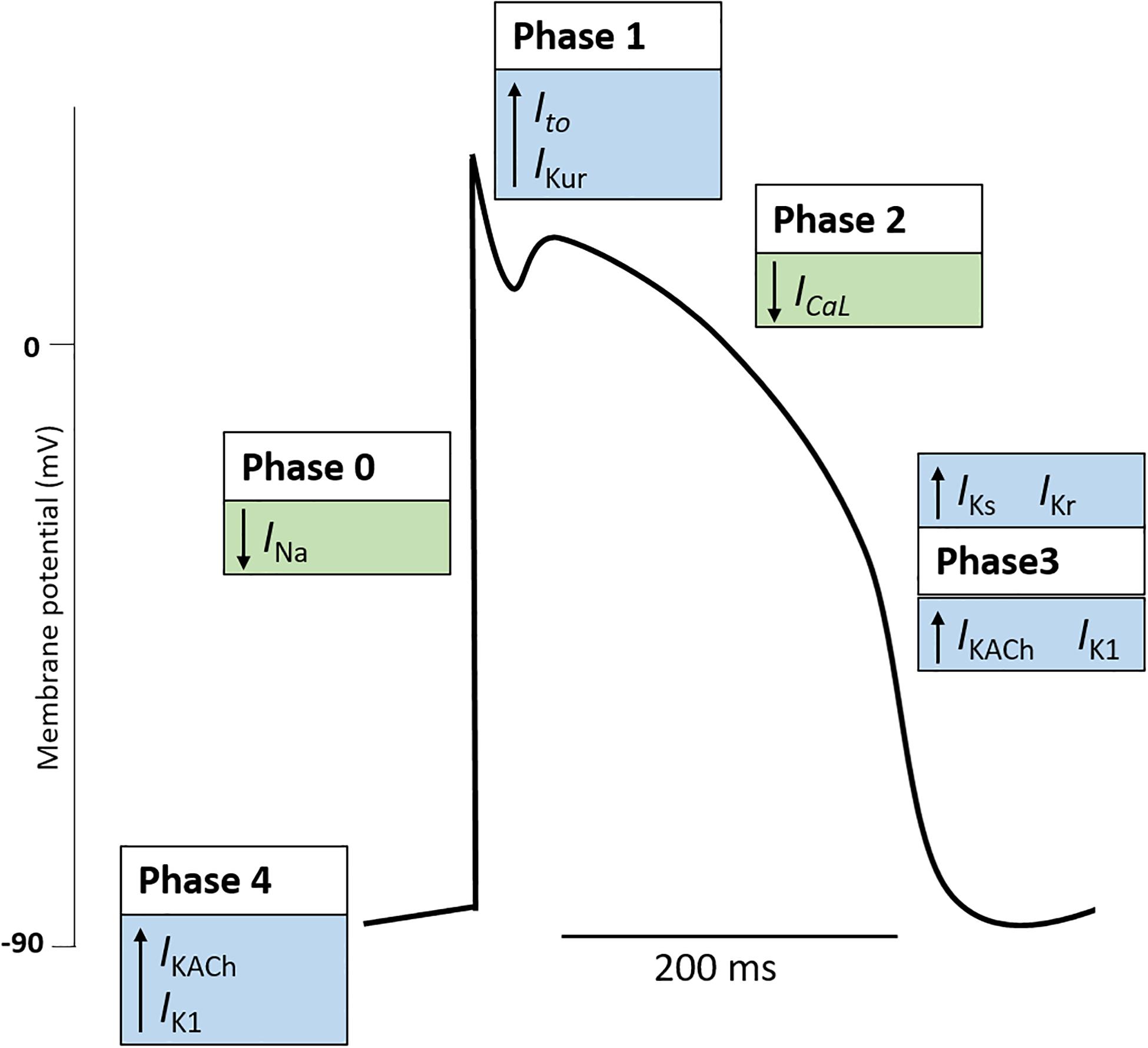
Figure 1. The cardiac action potential. The cardiac action potential represents the electrical activity of a cardiomyocyte during cardiac contraction and is governed by changes in outward (blue boxes) and inward (green boxes) cardiac ion currents. Generally, the cardiac action potential is divided into five Phases. (Phase 4: Resting) The resting membrane potential is stable (∼–90 mV) and maintained by outward leak of K+ (IKACh, IK1). Here the Na+ and Ca+ channels are closed. (Phase 0: Upstroke) An action potential at a neighboring cardiomyocyte initiates a rise in membrane potential. When the membrane potential reaches the threshold potential (∼70 mV), Na+ channels open, resulting in a rapid influx of Na+ and the generation on the inward Na+ current (INa). Na+ channels become inactivated soon after opening (Phase 1: Early repolarization). Some K+ channels transiently open causing an outward K+ current (Ito, IKur). This results in a period of rapid repolarization. (Phase 2: Plateau phase) L-type Ca2+ channels are open to create a small, constant inward Ca2+ current (ICaL) which triggers excitation contraction coupling resulting in cardiomyocyte contraction. K+ currents remain active, although the two currents are electrically balanced and maintain a plateau. (Phase 3: Repolarization) Ca2+ channels become gradually inactivated and the outflow of K+ exceeds the Ca2+ inflow. K+ currents (IKs, IKr, IKACh, IK1) repolarize the cell and the resting membrane potential is restored. Note, the figure represents the typical ventricular cardiac action potential. The action potential of atrial cells and pacemaker cells (those found in the sinus node, atrioventricular node, and Purkinje fibers) differ. K+, potassium; Na+, sodium; Ca2+, calcium; IKACh, muscarinic-gated K+ current; IK1, inward rectifier K+ current; INa, Na+ current; Ito, transient outward K+ current; IKur, delayed rectifier K+ current; ICaL, L-type Ca2+ current; IKs, delayed rectifier (slow) K+ current; Ikr, delayed rectifier (fast) K+ current.
In the heart, Ca2+ has an indispensable role in cardiomyocyte ECC; in addition, as a signaling molecule, Ca2+ also acts as an intracellular second messenger in cardiomyocytes and other cell types. ECC results in cardiac contraction and helps to maintain diastolic and systolic cardiac function (Lou et al., 2012). During ECC, four steps involving Ca2+ trigger cardiomyocyte contraction: (1) membrane depolarization causes a rapid influx of Ca2+ through the LTCC located on transverse (T)-tubules, which results in an intracellular Ca2+ gradient in the junctional zone; (2) Ca2+ diffusion activates RyR located on the SR resulting in Ca2+ sparks and an amplification of the Ca2+ signal in a process termed Ca2+-induced Ca2+ release; (3) Ca2+ released via RyR from internal SR stores binds to troponin-C to cause contraction through actin-myosin filament crossbridge formation; (4) Ca2+ is transported out of the cardiomyocyte through NCX and PMCA or back into internal stores through SERCA and MCU (Bers, 2002; Lou et al., 2012) (Figure 2). The function of SERCA is tightly regulated by PLN. In its dephosphorylated form, PLN inhibits SERCA2a but when its phosphorylated at Ser10, Ser16 or Thr17, the inhibition of SERCA2 is relieved (Mattiazzi et al., 2005). Such is the importance of PLN-mediated SERCA2a regulation that PLN ablation induces a hypercontractile cardiac phenotype (Luo et al., 1994). Ca2+ homeostasis is also maintained through additional Ca2+ channels located on the plasma membrane (Figure 2). The transient receptor potential channels (TRP) are a large family of non-selective cation permeable channels which, depending on the family, permit the influx of Ca2+, along with Mg2+, Zn2+, and Na+(Falcon et al., 2019). The TRP family can be further divided into subfamilies, including TRP-canonical (TRPC), TRP vallinoid-related (TRPV), and TRP melastatin-related (TRPM) (Freichel et al., 2017). The ECC Ca2+ cycle is tightly regulated by protein kinases and accessory proteins which modify the activity and expression of ECC Ca2+ handling proteins. Functional changes to proteins involved in ECC and Ca2+ homeostasis can impair contractility and contribute to cardiac dysfunction. Furthermore, changes to Ca2+ handling can also impair cardiac conductivity, causing fatal arrhythmias. Altered Ca2+ homeostasis can result in abnormal automaticity, triggered activity, and can provide a substrate for re-entry arrhythmias (Landstrom et al., 2017). The contributions these Ca2+ handling proteins make to the pathophysiology of cardiac hypertrophy, HF, and cardiac arrhythmias are discussed in detail later in this review.
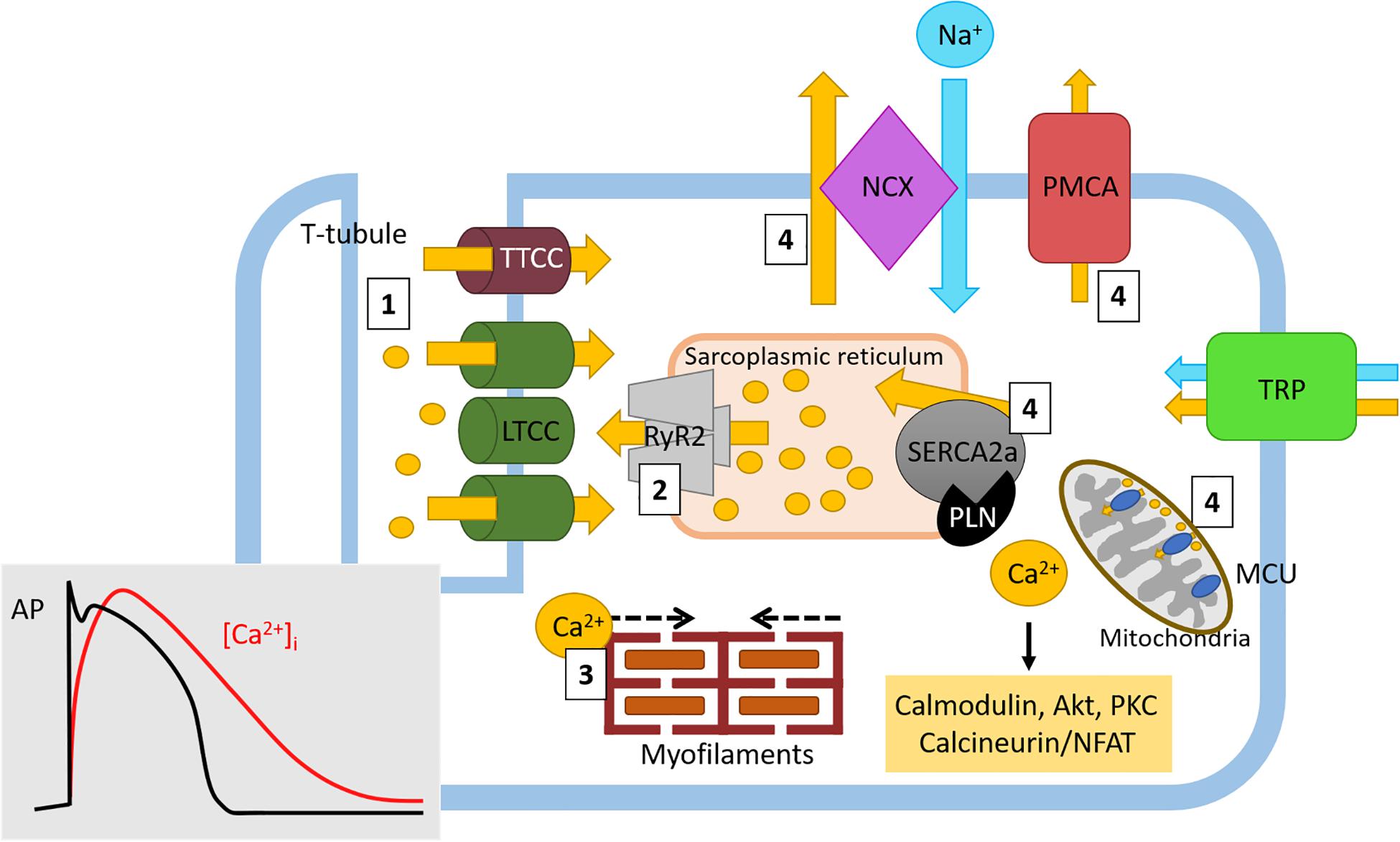
Figure 2. Physiological Ca2+ cycling in cardiomyocytes. Ca2+ cycling in cardiomyocytes generates cardiac contraction through excitation-contraction coupling. Briefly, (1) Ca2+ (yellow circles and arrows) rapidly enters the cell through LTCC where it causes (2) Ca2+-induced Ca2+ release from Ca2+ stores in the sarcoplasmic reticulum. During these early stages of ECC, intracellular Ca2+ ([Ca2+]i) levels rapidly increase (red line, insert) following the onset of the cardiac action potential and cardiomyocyte depolarization (and black line, insert). (3) The amplified Ca2+ signal then induces actin-myosin myofilament crossbridge formation, resulting in cardiac contraction. Following this action, as the cardiac action potential enters the plateau phase (black line), the inward [Ca2+]i levels begin to slowly decrease (red line). (4) Ca2+ is either returned to internal stores through SR-located SERCA2a and mitochondrial MCU or to the extracellular space through NCX (in exchange for 3Na+, blue circle and arrow) and PMCA. Here, as cardiac repolarization occurs (black line) the levels of [Ca2+]i return to baseline (red line) in order to return membrane polarization to baseline. Free Ca2+ can also drive intracellular signaling (yellow box) which can influence gene transcription. Ca2+ homeostasis is also maintained by the TRP channel family of proteins. Ca2+, calcium; Na+, sodium; LTCC, L-type Ca2+ channel; TTCC, T-type Ca2+ channel; NCX, sodium calcium exchanger; PMCA, plasma membrane Ca2+ ATPase; TRP, transient receptor potential (canonical, vallinoid-related, melastatin-related), RyR, ryanodine receptor; SERCA, sarco/endoplasmic reticulum Ca2+-ATPase; PLN, phospholamban; MCU, mitochondrial Ca2+ uniporter.
Current Therapeutic Approaches Targeting HF and Cardiac Arrhythmia and Their Impact on Ca2+-Handling Proteins
Treatment of HF and cardiac arrhythmias can be surgical (including implantation of a pacemaker or implantable cardioverter defibrillator) and/or pharmacological. Primary drug-based therapies for these disorders include targeting (1) the RAAS signaling system, (2) β-adrenergic hyperactivation, and (3) aberrant ion channel activation (Ca2+, Na+, and K+). As the focus of this review concerns how Ca2+ handling proteins present a novel therapeutic approach, we discuss the current pharmacological therapies mentioned above in brief, including any known impact they have on Ca2+ handling in the heart.
The Renin–Angiotensin–Aldosterone System
Renin–angiotensin–aldosterone signaling is elevated in patients with HF and reduced ejection fraction (HFrEF, ejection fraction < 40%) (Watkins et al., 1976) and in advanced HF, the levels of plasma renin activity and aldosterone are increased (Dzau et al., 1981). Furthermore, aldosterone secretion is enhanced in response to ANG-II signaling and its receptor is upregulated in cardiomyocytes from HFrEF patients (Yoshida et al., 2005). Aldosterone alone is pro-hypertrophic, pro-fibrotic and pro-inflammatory (Pinilla-Vera et al., 2019). Increased neurohormonal signaling can activate the MAPK which, in turn, upregulates pro-hypertrophic gene transcription (Zhang et al., 2003; Nakamura and Sadoshima, 2018). The MAPK signaling cascade comprises four main ‘typical’ branches; ERK1/2, JNK, p38 and ERK5, all with distinct upstream regulators (Rose et al., 2010) and activation of the ERK1/2 signaling cascade may determine whether concentric or eccentric cardiomyocyte growth and remodeling occurs (Bueno et al., 2000; Lorenz et al., 2009; Kehat et al., 2011). Activation of RAAS can also promote cardiac arrhythmias including AF and ventricular tachyarrhythmias, partly due to the formation of a pro-arrhythmia substrate in cardiac hypertrophy and HF. In addition, RAAS is also known to directly affect cardiac ion channels. Angiotensin and aldosterone have been shown to attenuate the transient outward K+ current (Ito) and enhance ICaL, impacting AP duration and repolarization. Whilst oxidative stress triggered by RAAS activation is reported to affect cardiac ion currents and several Ca2+ handling proteins (Iravanian and Dudley, 2008).
The maladaptive effects associated with over-activation of the RAAS is one of main focuses of current therapeutics; both ACEI and ARBs are prescribed to patients with HF to help improve cardiac function and survival (McMurray, 2004; Demers et al., 2005). Several clinical trials have shown that ACEI and ARBs reduce mortality in patients with HFrEF (Kjekshus et al., 1992; Maggioni et al., 2002; McMurray et al., 2003). In addition, further inhibition of RAAS is achieved through aldosterone antagonism. Aldosterone receptor blockade with an agent such as eplerenone has been shown to reduce the risk of death and hospitalization events in patients with HFrEF (Pitt et al., 2003; Zannad et al., 2011). However, the absolute beneficial use of aldosterone agonists in patients with HF and preserved ejection fraction (HFpEF, ejection fraction > 50%) is still yet to be determined (Pitt et al., 2014). Studies have also aimed to dissect the use of RAAS inhibitors in patients with cardiac arrhythmias (Iravanian and Dudley, 2008). ACEI and ARBs have been shown to reduce the recurrence rate in patients with existing AF (Madrid et al., 2002; Ueng et al., 2003), and reduce the incidence of new onset AF in hypertensive (Wachtell et al., 2005)- and post-MI (Pedersen et al., 1999) patients. Treatment with ACEI has also been associated with a reduced risk of sudden cardiac death in HF patients (Heart Outcomes Prevention Evaluation Study Investigator, Yusuf et al., 2000).
The β-Adrenergic Pathway
In HF, elevated sympathetic activity increases blood catecholamines; an increase which inversely correlates with survival (Lymperopoulos et al., 2013). The initial elevation in sympathetic activation is beneficial as cardiac contractility is enhanced to maintain function; however, chronic activation of β-adrenergic receptors (β-AR) promotes cardiac dysfunction through increased cardiomyocyte hypertrophy (Lohse et al., 2003). Catecholamine binding to β-AR activates AC and subsequently leads to an increase in cAMP levels; cAMP activates PKA which phosphorylates proteins associated with Ca2+ handling, including LTCC (Schroder et al., 1998), RyR2 (Wehrens et al., 2006), and PLN (Antos et al., 2001; Nakamura and Sadoshima, 2018), together culminating in increased levels of cytosolic Ca2+ (Port and Bristow, 2001; Nakamura and Sadoshima, 2018). In addition to phosphorylation of key Ca2+ handling proteins, β-AR signaling also results in the s-nitrosylation of many Ca2+ handling proteins in cardiomyocytes, such as PLN and cardiac troponin C (Irie et al., 2015). Furthermore, activation of β-AR is the primary mechanism underlying sympathetic control of heart rhythm, with many of the cardiac ionic currents responsive to β-adrenergic stimulation (Gardner et al., 2016). Unsurprisingly, enhanced adrenergic activity has been identified in arrhythmic conditions, including AF, and results in enhanced automaticity and triggered activity (Workman, 2010; Antzelevitch and Burashnikov, 2011).
Therapeutic targeting of this excessive β-adrenergic response is achieved using β-blockers. β-blockers are deployed to improve cardiac function, reduce incidence of arrhythmias, and decrease mortality associated with HF by diminishing the hyperactivity of the β-adrenergic pathway. Improvements in mortality rates in patients with HFrEF have been recorded, irrespective of the type of β-blocker (Chatterjee et al., 2013; Pinilla-Vera et al., 2019), and large randomized trials have provided further evidence of the advantageous effects of single β-blockers agents in the treatment of HFrEF (CIBIS-II Investigators and Committees, 1999; Tepper, 1999; Packer et al., 2002). Furthermore, a meta-analysis of all clinical trials involving the use of β-blockers in HFpEF patients suggested the use of β-blockers may reduce mortality by 22% (Fukuta et al., 2017). Interestingly, through their mechanism of action, β-blockers also inhibit PKA hyperphosphorylation of the RyR2 channel in both human and animal models of HF (Reiken et al., 2001, 2003); the proposed mechanism by which β-blockers improve cardiac function in HF is by preventing PKA hyperphosphorylation of RyR2 Ser2808 and subsequent receptor destabilization (Shan et al., 2010). β-blockers also display multiple antiarrhythmic effects including a lowering of heart rate, a decrease in SAN/AVN automaticity and re-entry events, and a reduction in afterdepolarizations. Notably, β-blockers are used for the treatment of sinus and supraventricular tachycardias and in rate control of AF and ventricular tachyarrhythmias (Lei et al., 2018). In addition, β-blockers are also used to treat some patients with LQTS, thus reducing the risk of adverse cardiac events; although it is reported that some β-blockers are more effective than others (Chockalingam et al., 2012; Abu-Zeitone et al., 2014). Patients with CPVT are prescribed β1/β2-blockers such as nadolol which, when compared to β1-selective blockers, reduces the severity and incidence of ventricular arrhythmias (Leren et al., 2016).
Ion Channel Blockers; Ca2+, Na+, and K+
The voltage activated LTCC is a multi-subunit protein which, under basal conditions, provides the primary entry point for rapid Ca2+ influx. During HF, increased phosphorylation of LTCCs may result in redistribution of LTCCs and increased channel open probability (Schroder et al., 1998; Sanchez-Alonso et al., 2016) which can lead to increased Ca2+ influx, a phenomenon implicated in both hypertrophy and HF (Richard et al., 1998). Changes in LTCC dynamics have also been associated with the development of arrhythmias, and numerous LTCC mutations have been identified in primary arrhythmic disorders such as LQTS and Brugada syndrome (Grant, 2009). The LTCC is the primary target of Ca2+ channel blockers which are currently extensively used to treat hypertension and certain arrhythmic disorders, but are used with caution in HF. Inhibiting the LTCC with amlodipine prevents adverse cardiac remodeling in a mouse model of TAC-induced pressure overload hypertrophy (Liao et al., 2005). However, cardiac-specific knockdown of LTCC has the opposing effect and results in pathological hypertrophy partly due to a compensatory increase in Ca2+ leak from the SR via RyR2 (Goonasekera et al., 2012).
Ca2+ channel blockers (non-dihydropyridines and dihydropyridines) are generally not prescribed to patients with HFrEF, except for amlodipine. Due to their negative inotropic effects, clinical studies have demonstrated that non-dihydropyridines increase mortality rates and incidence of late-onset HF (The Danish Study Group on Verapamil in Myocardial Infarction, 1990; Goldstein et al., 1991) and should be avoided to prevent further depression of cardiac function. However, amlodipine (a dihydropyridine) does not affect mortality so can be used to treat HFrEF patients with angina and/or hypertension (Packer et al., 1996). On the other hand, both classes of Ca2+ channel blockers have no adverse effects on mortality or HF hospitalization in patients with HFpEF (Patel et al., 2014); although, non-dihydropyridines tend only to be considered when β-blockers alone are unsuccessful (Godfraind, 2017).
Ca2+ channel blockers are currently approved antiarrhythmic agents used to treat arrhythmias arising from SAN and AVN dysfunction; the AP generated by these areas of the heart are strongly dependent on Ca2+ entry (Lei et al., 2018). The non-dihydropyridines Ca2+ channel blockers used for the treatment of arrhythmias (supraventricular arrhythmias, ventricular tachycardias occurring in the absence of structural heart disease, and AF) include phenylalkylamines and benzothiazepines (for example, verapamil, and diltiazem, respectively) (January et al., 2014; Lei et al., 2018). However, as noted above, non-dihydropyridines should not be used in patients with decompensated HF. In addition, non-dihydropyridines should be avoided in patients with AF combined with pre-excitation (a condition in which the ventricles activate too early) as treatment may increase the risk of ventricular fibrillation (January et al., 2014). There is evidence that combining Ca2+ channel blockers with other pharmacological approaches can enhance the treatment of arrhythmic conditions. For example, in patients with CPVT, the addition verapamil to existing β-blocker therapy was shown to reduce the incidence of exercise-induced arrhythmias when compared to β-blocker therapy alone (Rosso et al., 2007).
Na+ channel blockers target the voltage-gated Na+ channel, Nav1.5, which drives the inward Na+ current responsible for the rapid depolarization at the start of the cardiac AP (Lei et al., 2018). The aim of Na+ channel blockers is therefore to reduce the rate of the AP upstroke and reduce AP conduction in the atria, ventricle, and Purkinje fiber tissue (where Nav1.5 is preferentially expressed). Class I Na+ channel blockers are further subclassified based on their molecular properties and interaction with the Nav1.5 (Lei et al., 2018; Li et al., 2019). Class Ia (quinidine, ajmaline, disopyramide) and Ib (lidocaine, mexiletine) Na+ channel blockers are used to treat patients with sustained ventricular tachycardia and ventricular fibrillation. In addition, quinidine has been indicated for short QT syndrome and Brugada syndrome and mexiletine suggested for the treatment of premature ventricular complexes, LQT3, and ventricular arrhythmias after MI (Al-Khatib et al., 2018; Lei et al., 2018). Class Ic agents (flecainide, propafenone) are used for ventricular tachycardia, premature ventricular complexes, and AF, with flecainide also having a role in the treatment of CPVT (January et al., 2014; Al-Khatib et al., 2018). However, Class Ic agents should not be used in patients with underlying structural heart disease (January et al., 2014). Ranolazine, a Class Id agent, is used to treat ventricular tachycardia and differs from Class Ia-Ic agents by inhibiting the late Na+ current (Al-Khatib et al., 2018; Lei et al., 2018). Despite Na+ channel blockers being identified as an effective antiarrhythmic treatment; they have also been shown to be proarrhythmic. The Cardiac Arrhythmia Suppression Trial (CAST) in the late 1980s studied the effect of Na+ channel blockers on patients with ventricular ectopic activity up to 2 years following a MI. The study revealed a higher rate of death in patients receiving flecainide or encainide compared to the placebo group (Cardiac Arrhythmia Suppression Trial I., 1989). Na+ channel blockers are therefore generally limited to patients who do not have structural heart disease.
The Class III K+ channel blockers work by binding to K+ channels involved in Phase 3 repolarization, and thus delay repolarization and increase AP duration (Lei et al., 2018). Class IIIa K+ blockers can be divided based on their K+ channel target. Non-selective K+ channel blockers (ambasilide, amiodarone, dronedarone) and Kv11.1 blockers (dofetilide, ibutilide, sotalol) have been indicated for the treatment of ventricular tachycardia in patients with structural heart disease or with remote MI, tachyarrhythmias in patients with Wolff-Parkinson-White syndrome, rate control in AF [with amiodarone the most effective antiarrhythmic drug for maintenance of sinus rhythm in patients with persistent or paroxysmal AF (January et al., 2014)], ventricular fibrillation and premature ventricular contraction, and supraventricular tachyarrhythmias (January et al., 2014; Al-Khatib et al., 2018; Lei et al., 2018). Another type of K+ channel blocker which targets Kv1.5 (vernakalant) has been suggested for the treatment of AF (Lei et al., 2018). Similar to Na+ channel blockers, K+ blockers can exhibit proarrhythmic effects. QT interval prolongation caused by K+ channel blockers has been shown to predispose patients to ventricular arrhythmias and TdP (McCauley et al., 2016). Due to the AP prolongation effect, K+ channel blockers should not be given to patients with LQTS, those being treated with QT-prolonging drugs, or with comorbidities associated with an increased risk for TdP (Dan et al., 2018). In addition, co-administration of amiodarone with other cardiac therapeutics including β-blockers, can result in bradycardia and atrioventricular block (Dan et al., 2018). As well as K+ channel blockers, Class IIIb drugs are metabolically dependent K+ channel openers and target Kir6.2 channels (although these have not been indicated in the treatment of arrhythmias) (Lei et al., 2018). A recent updated classification determined a Class IIIc group made up of transmitter dependent K+ channel blockers with BMS 914392 currently under review for the management of AF (Lei et al., 2018).
Pathological Cardiac Hypertrophy and Heart Failure
Cardiac hypertrophy occurs under both physiological and pathological conditions. Hypertrophy itself is an adaptive cardiac response which arises due to either pressure- or volume-cardiac overload. Typically, in physiological hypertrophy, the increase in cardiomyocyte size is initially due to the addition of sarcomeres in parallel (eccentric hypertrophy) which increases both ventricular volume and relative wall thickness, resulting in increased contractility which preserves cardiac output (Nakamura and Sadoshima, 2018). Conversely, in pathological hypertrophy cardiomyocytes are added in series (concentric hypertrophy) in order to compensate for the increased cardiac stress, this type of hypertrophy results in maintained or reduced ventricle volume but an increased relative wall thickness (Sawada and Kawamura, 1991; Lazzeroni et al., 2016; Nakamura and Sadoshima, 2018). Furthermore, persistent maladaptive remodeling can eventually lead to ventricular dilatation (eccentric hypertrophy), contractile dysfunction and impaired cardiac output (Sawada and Kawamura, 1991; Lazzeroni et al., 2016; Nakamura and Sadoshima, 2018).
It is now widely believed that each form of hypertrophy is regulated by distinct molecular mechanisms that drive the differing cardiac phenotypes and overall prognosis. Despite the activation of initial compensatory hypertrophic mechanisms, maladaptive remodeling occurs when the following processes occur: cell death (apoptosis, necrosis, autophagy), deposition of fibrosis throughout the myocardium, impaired mitochondrial function and regulation, dysregulation of Ca2+-handling proteins, re-expression of fetal genes, changes to sarcomere structure and rarefaction of the cardiac microvasculature (Nakamura and Sadoshima, 2018). Chronic maladaptive remodeling witnessed during the pathological response to hypertrophy often develops into HF with the diagnosis of HF itself rapidly becoming a universal healthcare and financial burden. HF is an end-stage CVD which frequently occurs secondary to many different aetiologies (Ziaeian and Fonarow, 2016). Physiologically, HF can be defined as (1) insufficient cardiac output to meet metabolic demands or (2) adequate cardiac output with increased left ventricular filling pressure through activation of compensatory neurohormonal pathways (Ponikowski et al., 2016). HF can be further classified into one of three subtypes: HFpEF, HFrEF or HFmrEF) (Ponikowski et al., 2016; Rastogi et al., 2017). These subclassifications are divided based on quantitation of ejection fraction; HFrEF < 40%, HFmrEF > 40% but < 50% and HFpEF > 50% (Ponikowski et al., 2016; Ziaeian and Fonarow, 2016; Rastogi et al., 2017).
The presence of altered Ca2+ transients in HF has been well-documented since the late 1980s. These early studies were amongst the first to show that cardiomyocytes isolated from the failing human heart exhibit reduced Ca2+ transient amplitudes, fewer actin-myosin crossbridge formations, a slower rate of Ca2+ removal and increased resting intracellular Ca2+ when compared to healthy cardiomyocytes (Gwathmey et al., 1987, 1991; Beuckelmann et al., 1992; Hasenfuss et al., 1993). In broad terms, the failing cardiomyocyte shows impaired Ca2+ cycling, Ca2+ release from the SR and Ca2+ removal. Therefore, altering Ca2+ in HF through modulation of Ca2+ handling proteins has long been investigated as a therapeutic target.
Calcium Handling Proteins in Cardiac Hypertrophy and HF and Their Potential as Novel Therapeutic Targets
The National Institute of Clinical Excellence (NICE, United Kingdom) and other regulatory bodies such as the European Society of Cardiology recommends ACEIs and β-blockers as first-line treatment for HF (Al-Mohammad et al., 2010; Ponikowski et al., 2016); however, dysregulation of Ca2+ handling proteins occurs exclusively during pathological hypertrophy (and not physiological hypertrophy) which suggests altered Ca2+ cycling and signaling precedes Ca2+ dysregulation observed in HF. The following section of this review will focus on how proteins associated with Ca2+-handling, encompassing Ca2+ signaling proteins and Ca2+ channels and pumps, are altered in cardiac hypertrophy and HF.
Ca2+ Homeostasis and Signaling in Hypertrophy and HF
Given the importance of Ca2+-dependent signaling pathways in the development of hypertrophy and HF, there is scope for developing therapies which target their aberrant expression and activity. Three such potential targets are calmodulin (CaM), calcineurin and CaMKII which are all activated following the release of Ca2+ from internal stores. CaMKII signaling is upregulated in human HF (Anderson, 2009). Evidence from animal studies suggests that CaMKII-δ is important in the cardiac stress response as CaMKII-δ null mice show no overt cardiac phenotype under physiological conditions but have an attenuated response to pressure-overload induced hypertrophy (Backs et al., 2009; Ling et al., 2009). CaMKII-δ may influence cardiac hypertrophy through two distinct mechanisms which are mediated by different splice variants; CaMKII-δC regulates phosphorylation of RyR and PLN resulting in increased Ca2+ spark frequency and decreased SR Ca2+ content whereas both CaMKII-δC and CaMKII-δB variants mediate pro-hypertrophic gene transcription (Zhang et al., 2007). Despite this data to suggest CaMKII plays an important role in HF, no clinically available compounds are currently in clinical trials. However, a recently developed, novel ATP-competitive CaMKII-δ and CaMKII-γ inhibitor, RA306, has shown potential in animal studies (Beauverger et al., 2020) (Figure 3 and Table 1). RA306 prevented phosphorylation of PLN (Thr17) in vitro and in vivo following stimulation with isoproterenol (Beauverger et al., 2020). Furthermore, twice-daily RA306 treatment was sufficient to increase ejection fraction in a mouse model of dilated cardiomyopathy; importantly, the study used aged mice with advanced cardiac disease to demonstrate the ‘curative’ ability of RA306 (Beauverger et al., 2020).
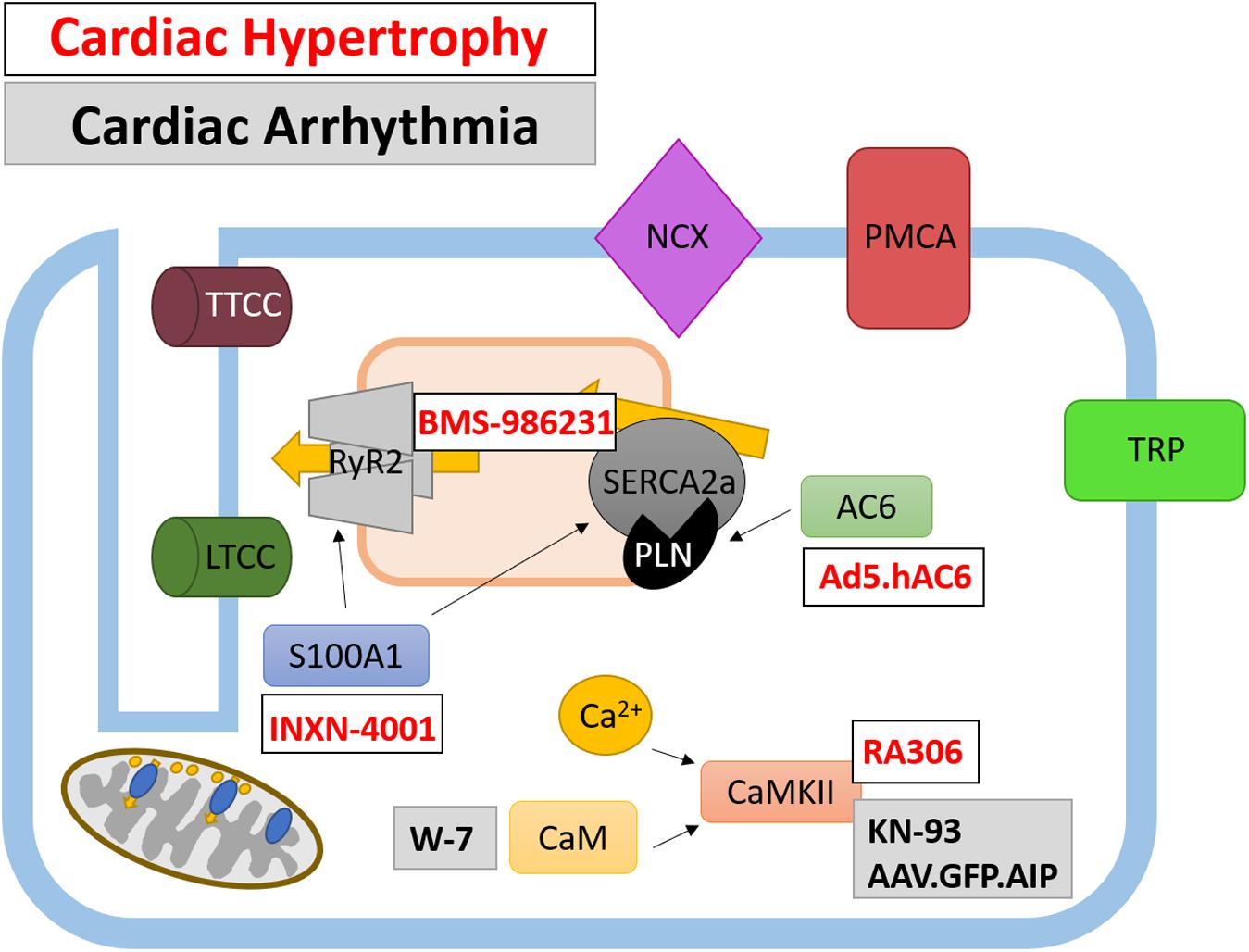
Figure 3. Therapeutically targeted Ca2+-handling proteins and their function in cardiomyocytes. Ca2+ handling proteins are extensively involved in intracellular signaling and the regulation of Ca2+-channels and pumps, as such their modulation presents a different avenue for therapeutic targeting. Both current and potential therapeutics are shown for cardiac hypertrophy (white box, red text) and cardiac arrhythmias (gray box, black text).
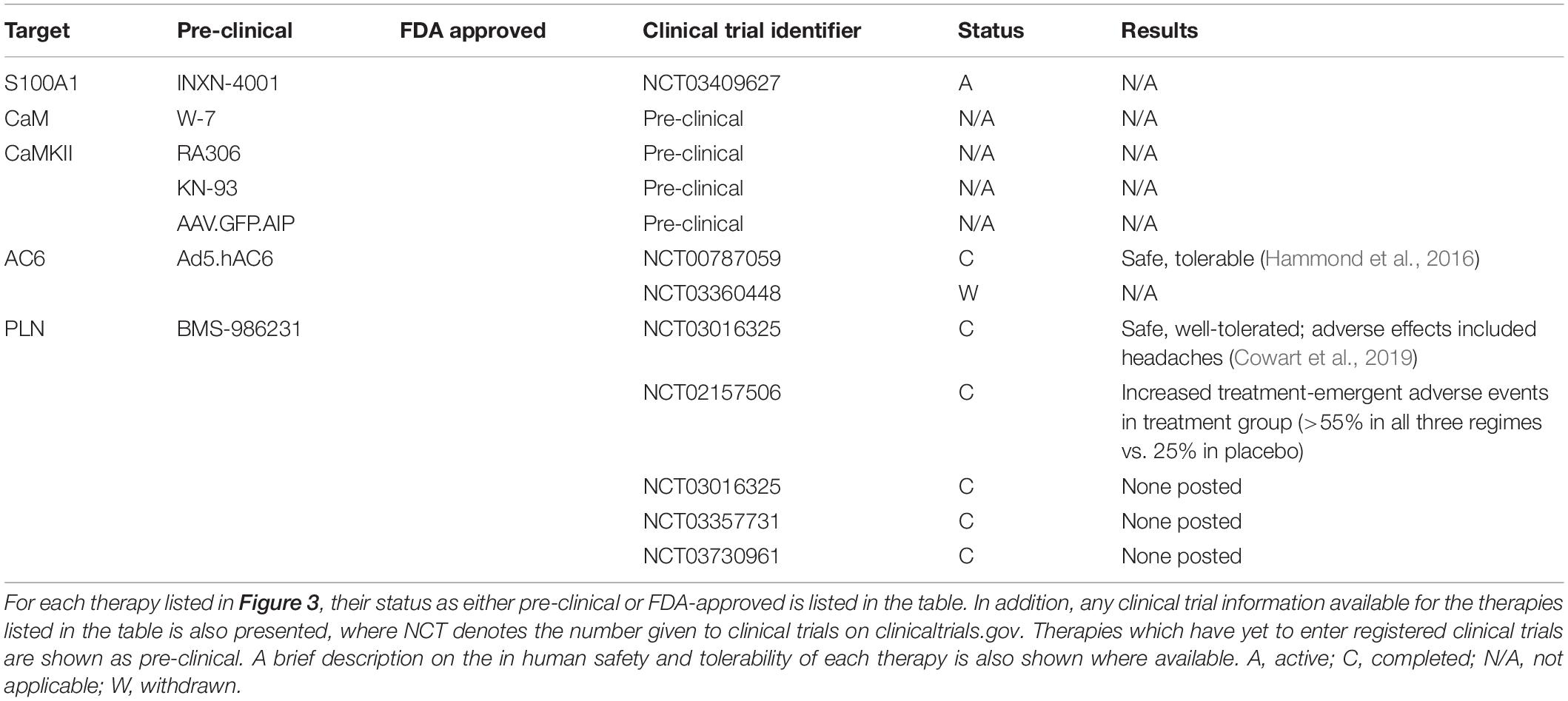
Table 1. Therapeutic targeting of Ca2+-handling proteins in cardiac hypertrophy, HF and arrhythmia.
The use of gene therapy to treat CVD has garnered much attention in recent years and has been used as a tool to alter expression of a wide variety of Ca2+-handling proteins. Recent work has been undertaken to trial a triple gene plasmid construct INXN-4001 in patients with an outpatient left ventricular assist device [clinicaltrials.gov Identifier NCT03409627] (Figure 3 and Table 1). Plasmid INXN-4001 contains human VEGF165, S100A1 and stromal cell-derived factor 1; expression of these three proteins has been designed to target fibrosis, angiogenesis and impaired cardiac contractility (Jaruga-Killeen et al., 2019). To date there are no reported adverse cardiac-specific safety effects after one infusion of INXN-4001 (Jaruga-Killeen et al., 2019). Of interest to this review is the expression of constitutively active S100A1 protein within the INXN-4001 plasmid. S100A1 is a Ca2+ binding protein which controls Ca2+-dependent networks in cardiomyocytes through, for example, modulation of RyR2 and SERCA2a activity (Ritterhoff and Most, 2012). S100A1 protein levels are reduced in cardiomyocytes from both human and animal model-derived failing myocardium and levels of reduced S100A1 are associated with poorer contractile function (Du et al., 2002; Most et al., 2006). Adenoviral-based delivery of S100A1 is able to rescue cardiac function in mouse, rat, and pig models of MI-induced HF (Most et al., 2004; Pleger et al., 2005, 2007, 2011; Kraus et al., 2009). Importantly, S100A1 gene transfer alone can restore Ca2+ homeostasis in acute and chronic models of HF (Most et al., 2004; Pleger et al., 2005, 2007, 2011; Kraus et al., 2009). However, gene therapy using S100A1 alone has not yet been developed for use in clinical trials.
Gene therapy has also been utilized to overexpress AC6 (Kieserman et al., 2019) (Figure 3 and Table 1). The primary role of AC6 is to catalyze the conversion of ATP to cAMP; however, AC6 can also downregulate the expression of PLN, independently of cAMP concentration (Gao et al., 2004). Adenoviral transfer of AC6 (Ad.ACVI) in a porcine model of congestive HF successfully improved LV function (Lai et al., 2004). The promising outcome of Ad.ACVI treatment in a large animal models of HF led to its development for a first in man clinical trial (Ad5.hAC6, also known as RT-100), beginning in 2010 (Hammond et al., 2016; Penny et al., 2018). Results from this placebo-controlled, randomized clinical trial of 56 HFrEF patients confirmed the safety of Ad5.hAC6 [clinicaltrials.gov Identifier NCT00787059] (Hammond et al., 2016). Furthermore, patients receiving the two highest doses (out of five possible different doses) of Ad5.hAC6 showed an acute improvement in EF, assessed 4 weeks post-viral delivery (Hammond et al., 2016). The promising results from this Phase II clinical trial paved the way for the ‘FLOURISH’ trial, a Phase III follow-up [clinicaltrials.gov Identifier NCT03360448] (Penny et al., 2018). However, the FLOURISH study was withdrawn prior to patient recruitment owing to a change of clinical development plans. Therefore, the clinical benefit of Ad5.hAC6 for the treatment of HF still requires investigation in a large, multi-centre clinical trial.
Ca2+ Handling Proteins During Hypertrophy and HF; Ca2+ Channels, Pumps and Exchangers
LTCC
Currently, LTCC Ca2+ blockers are used with caution in patients with HF; however, there may be some therapeutic potential in targeting a subset of LTCC which have been shown to specifically localize to caveolae and do not present as ‘typical’ LTCC which reside on T-tubules (Makarewich et al., 2012). Within caveolae membrane invaginations, Ca2+ entry through LTCC may activate pro-hypertrophic calcineurin/NFAT signaling without contributing to ECC (Makarewich et al., 2012). Interestingly, the calcineurin/NFAT signaling axis only contributes to pathological and not physiological hypertrophy (Wilkins et al., 2004). Inhibition of LTCC with channel blocking Rem proteins specifically targeted to Cav-3 caveolae membranes attenuates the inward Ca2+ current and prevents pro-hypertrophic gene activation without affecting contractility of feline cardiomyocytes (Makarewich et al., 2012). However, these findings were not replicated a murine model of cardiac hypertrophy in vivo (Correll et al., 2017). Therefore, the direct contribution of LTCC to the development of cardiac hypertrophy remains inconclusive and requires further investigation in different pre-clinical models of HF.
SERCA2
In failing human cardiomyocytes and in animal models of hypertrophy and HF, the levels of SERCA2 are reduced (Komuro et al., 1989; Nagai et al., 1989; Mercadier et al., 1990; Lipskaia et al., 2014). Downregulation of SERCA2 and therefore a reduction in SERCA2 activity are concordantly associated with both systolic and diastolic dysfunction (Periasamy et al., 1999). The importance of SERCA2 signaling is evidenced by knockout mouse studies. Cardiomyocyte-specific deletion of SERCA2 in adult mice leads to diastolic dysfunction under basal conditions (Andersson et al., 2009). Furthermore, an important feature of Ca2+ dysregulation during cardiac hypertrophy is SR Ca2+ leak and at the molecular level, cardiomyocyte loss of SERCA2 results in changes to SR Ca2+ handling, characterized by slower cytosolic Ca2+ removal, reduced SR content and impaired Ca2+ transient magnitude (Andersson et al., 2009; Louch et al., 2010; Li et al., 2012). SERCA2 activity could also be affected in HF due to changes in the phosphorylation status of PLN (Schwinger et al., 1995; Mattiazzi and Kranias, 2014). The reduction in phosphorylation of PLN is partly due to increased activity of calcineurin and protein phosphatase 1, as shown in both human HF and animal models (Neumann et al., 1997; Munch et al., 2002). However, more recent data suggests direct post-transcriptional modifications of SERCA2a contribute to its decreased activity (Kranias and Hajjar, 2012). In HF, SERCA2 can undergo either ‘stimulatory’ modification by glutathionylation on C674 (Adachi et al., 2004) and/or SUMOylation on K480 and K585 (Kho et al., 2011), and ‘inhibitory’ acetylation of K492 (Gorski et al., 2019). These modifications to SERCA2a and its regulatory proteins provides more scope for enhancing SERCA2 activity in HF patients.
Restoring SERCA2a protein expression levels has been shown by numerous groups to improve cardiac metabolism, including a reduced incidence of arrhythmia and increased coronary blood flow (Hayward et al., 2015; Hulot et al., 2016). However, the benefits of administering SERCA2a were shown to be diminished in a myocardium with a limited energy supply (Pinz et al., 2011), suggesting increasing SERCA2 levels may only be beneficial in non-ischemic tissue. In humans, directly restoring SERCA2 levels has been achieved using gene transfer. Adeno-associated virus1.SERCA2a (AAV1.SERCA2a) gene transfer successfully restored the Ca2+ transient and contraction in isolated failing human cardiomyocytes (del Monte et al., 1999) and in animal models of HF, SERCA2a gene transfer was shown to increase survival and improve cardiac hemodynamics and function (del Monte et al., 2001, 2004; Kawase et al., 2008) (Figure 4 and Table 2). Given that SERCA2a gene transfer improved outcome in animal models of HF, AAV1.SERCA2a was administered in Phase I and Phase II clinical trials [ClincalTrials.gov identifiers NCT00534703 and NCT00454818] (Jessup et al., 2011; Zsebo et al., 2014; Greenberg et al., 2016). The results of the initial Phase I and IIa trials suggested that AAV1.SERCA2a treatment reduced the number of cardiac events in HF patients (Jaski et al., 2009; Jessup et al., 2011); however, larger Phase IIb clinical trials failed to show improvement in any primary or secondary endpoints, leading to their early termination (Hulot et al., 2017). The failure of these clinical trials suggests the dosage and delivery method of AAV1.SERCA2a may require optimizing (Hulot et al., 2016) [SERCA2a gene therapy in HF has been extensively reviewed elsewhere (Samuel et al., 2018)]. Despite the lack of promising results for AAV1.SERCA2a, clinical trials are underway for istaroxime, a dual functional luso-inotropic agent which both stimulates SERCA2a and inhibits Na+/K+ ATPase (Micheletti et al., 2002) (Figure 4 and Table 2). Istaroxime increases Ca2+ uptake into the SR by relieving PLN-mediated inhibition of SERCA2a through cAMP/PKA-depending mechanism (Ferrandi et al., 2013). In animal models of HF, intravenous istaroxime improved cardiac function without causing any arrhythmic events (Micheletti et al., 2007; Sabbah et al., 2007). Phase I-II clinical trials demonstrated the safety of istaroxime in HF patients and showed beneficial effects on cardiac contractility (Ghali et al., 2007). The follow-up Phase IIa clinical trial, ‘HORIZON-HF,’ showed a reduction in diastolic stiffness and increase in contractility in HFrEF patients receiving istaroxime when compared to placebo (Shah et al., 2009). Additional clinical trials for istaroxime are on-going [ClincalTrials.gov identifiers NCT02617446 and NCT02477449].
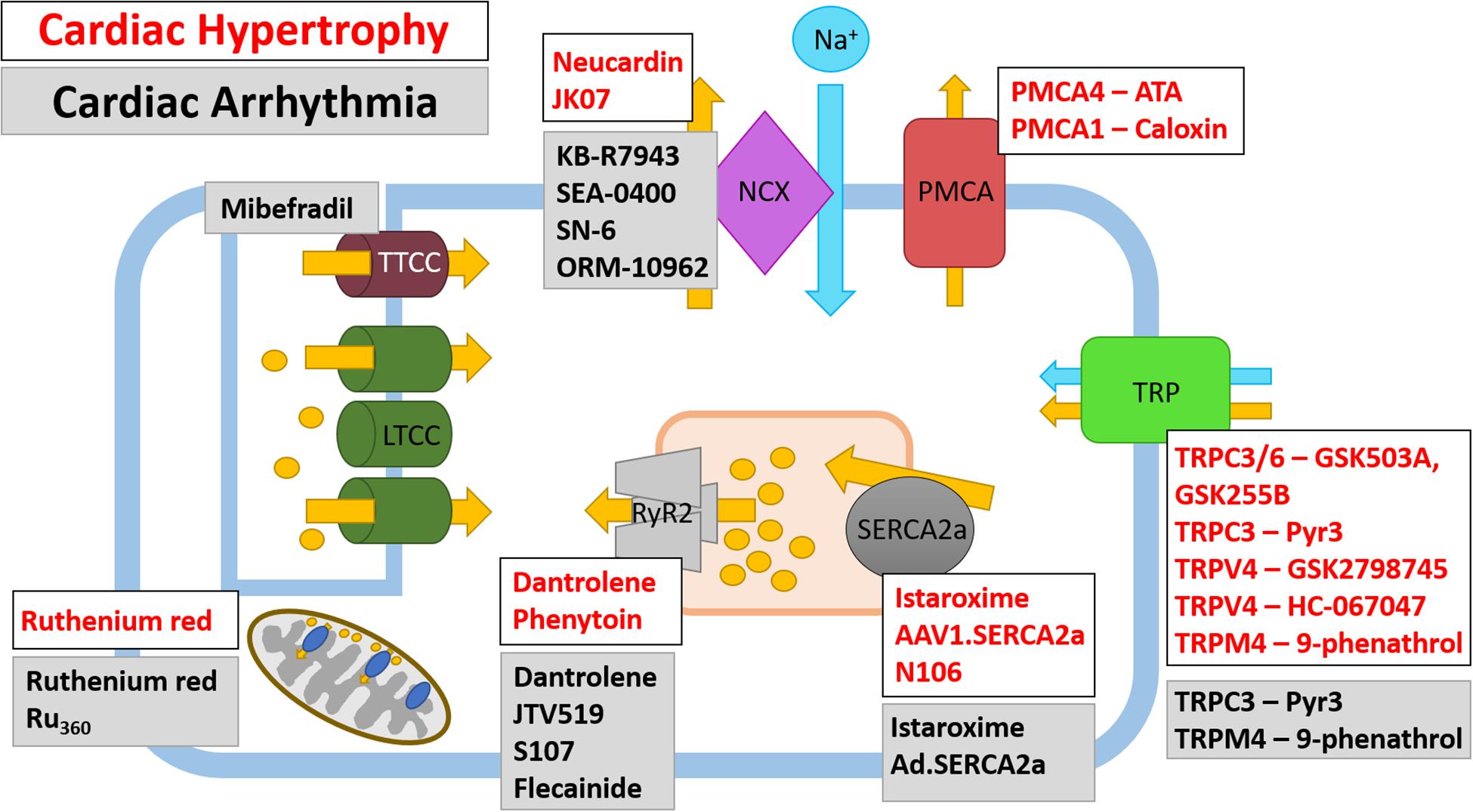
Figure 4. Therapeutic targeting of Ca2+ ion channels and pumps in cardiac hypertrophy and cardiac arrhythmia. Ca2+ ion channels and Ca2+ pumps have been the subject of pharmacological and gene therapy intervention; however, not all therapies have been translated from pre-clinical studies into human trials. Both current and potential therapeutics and their Ca2+ ion channel or Ca2+ pump target are shown for cardiac hypertrophy (white box, red text) and cardiac arrhythmias (gray box, black text), note there are some agents which have been tested in models of HF and arrhythmia. The LTCC are currently targeted by Ca2+ channel blockers which are prescribed to patients with CVD, as such, these are not shown on the schematic.
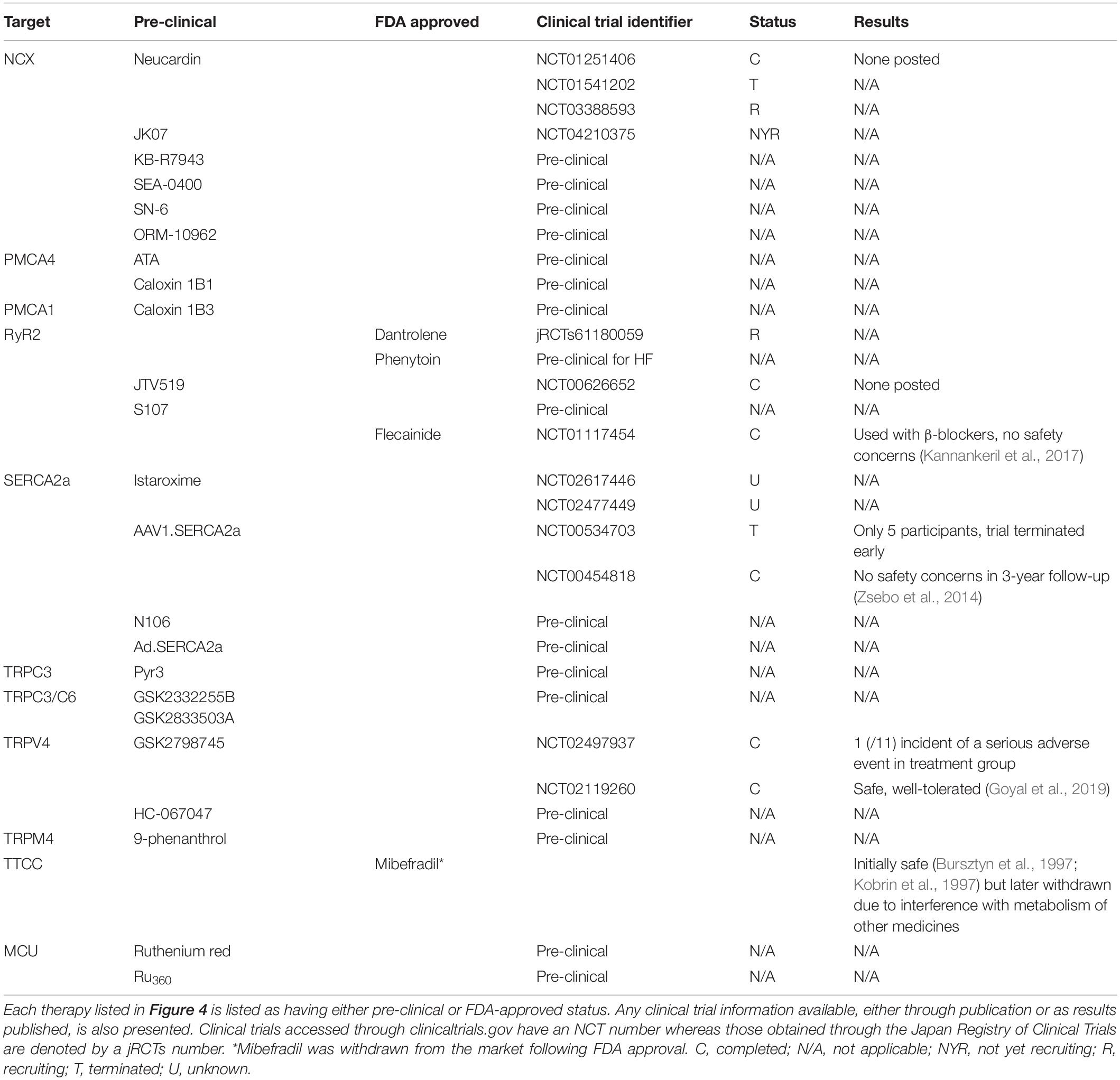
Table 2. Therapeutic targeting of Ca2+-channels and pumps in cardiac hypertrophy, HF and cardiac arrhythmia.
Focus for novel SERCA2 therapeutics has also extended to SERCA2 post-translational modifications and mediators of SERCA2 expression. Gene-transfer of SUMO1 and S100A1 which both promote SERCA2 activity were shown to improve cardiac function in rodent and porcine models of HF (Most et al., 2004; Kho et al., 2011; Pleger et al., 2011; Tilemann et al., 2013). Moreover, a small-molecule SUMO1 activator, termed N106, has recently been developed which, through activating the SUMO-activating enzyme, leads to the SUMOylation of SERCA2 (Kho et al., 2015). In addition, microRNA’s upregulated in human HF also provide a novel therapeutic target as they can be inhibited using anti-miR’s. One such microRNA, microRNA-25, is upregulated in human HF and application of anti-miR-25 following TAC improved survival and function by restoring SERCA2 activity (Wahlquist et al., 2014). However, none of these SERCA2 modifying mechanisms have been investigated in the clinical setting. SERCA2a and its regulator PLN can be modified by nitroxyl (HNO/NO–), the single electron reduced form of nitric oxide (Paolocci et al., 2001; Farah et al., 2018). HNO readily converts negatively charged thiol groups of proteins, such as cysteine, to disulfide residues; a feature which is reversible (Fukuto, 2019; Pinilla-Vera et al., 2019). HNO modifies cysteine residues on SERCA2a and PLN (and RyR2) which enhances Ca2+ cycling through reduced PLN-mediated SERCA2a inhibition (Froehlich et al., 2008; Farah et al., 2018). The original HNO donor used in proof-of-principal experiments, Angeli’s salt, is not stable at room temperature, therefore, first- and second-generation HNO donors have been developed with more favorable drug-like properties. One such second-generation HNO derivative to have undergone clinical testing is BMS-986231 (Figure 3 and Table 1). To date, there have been eight completed different clinical trials utilizing BMS-986231. Results from a Phase I clinical trial showed that BMS-986231 was well tolerated but that nearly half of healthy volunteers receiving BMS-986231 reported headaches; however, the authors noted that owing to BMS-986231 vasodilatory effects, the presence of headaches was not unexpected (Cowart et al., 2019) [clinicaltrials.gov identifier NCT03016325]. In a separate placebo-controlled, double-blind Phase IIa study, 34 patients were randomly assigned to receive one infusion of BMS-986231 (Tita et al., 2017) [clinicaltrials.gov identifier NCT02157506]. The results from this Phase IIa clinical trial also suggest BMS-986231 is safe and in HFrEF patients, exhibits a favorable hemodynamic profile (Tita et al., 2017). The success of these early Phase I and II clinical trials have led to the development of three further clinical trials to determine the effect of BMS-986231 on (1) symptomatic hypotension in HFrEF patients, (2) cardiac systolic and diastolic function in HFrEF patients, and (3) salt and water-handling in patients with HFrEF and HFmrEF (EF < 45%) [clinicaltrials.gov identifier(s) NCT03016325, NCT03357731, and NCT03730961, respectively] (Felker et al., 2019). To date, these studies have completed recruitment and are on-going.
NCX
Depending on the membrane potential and intracellular Ca2+ and Na+ gradients, NCX1 can act in ‘forward’ (Ca2+ ion out, 3 Na+ ions in) or ‘reverse’ (Ca2+ ion in, 3 Na+ ions out) mode. In a failing cardiomyocyte, NCX1 has been shown to operate in reverse mode which results in Ca2+ entry and a repolarizing current (Mattiello et al., 1998). Furthermore, elevated intracellular Ca2+ seen after I/R injury is caused by NCX1 operating in ‘reverse’ mode (Piper et al., 1996; Inserte et al., 2002; Imahashi et al., 2005). NCX1 is upregulated in human and murine models of HF (Hasenfuss et al., 1999; Menick et al., 2007) and over-expression of NCX is sufficient to impair cardiomyocyte function in vitro (Schillinger et al., 2000). However, as the function of NCX is also affected by Na+, and intracellular Na+ is increased in both human HF and models of HF (Bers and Despa, 2006), Ca2+ extrusion through NCX could be impaired due to Na+-loading observed in end stage HF (Louch et al., 2010; Li et al., 2012). Interestingly, following TAC there is an increase in NCX expression but no change in SERCA2a; however, when TAC progresses into HF, there is a downregulation in SERCA2 expression (Wang et al., 2001). Therefore, in HF there is a change in the NCX/SERCA2 ratio which has been implicated in impaired cardiac contraction and conduction (Pogwizd et al., 2001).
Historically, digoxin has been used to inhibit NCX; however, there is no improvement in mortality of patients receiving digoxin (Campbell and MacDonald, 2003; Goldhaber and Hamilton, 2010). It was recently observed that an increase in NCX1 seen in an animal model of HF could be attenuated by neuroglin-1β (Wang et al., 2019) (Figure 4). Neuroglin-1β has been extensively explored as a therapeutic option in HF and expression of recombinant human neuroglin-1β (rhNRG-1/also known as Neucardin) has undergone successful clinical trials; however, its full mechanism of action has not yet been determined (Galindo et al., 2014). Neuroglin-1β is a growth factor which, along with its receptors ErbB2-4, is critical for the cardiac response to stress (Lemmens et al., 2004; Galindo et al., 2014; Wang et al., 2019). Following pressure-overload induced cardiac hypertrophy, the expression of ErbB2 and B4 are increased during the cardiac compensation phase and reduced following the transition into HF (Rohrbach et al., 1999). Similarly, in the left ventricle of human failing myocardium, the expression of ErbB2 and B4 is reduced (Rohrbach et al., 2005). Animal models of hypertrophic- and ischemia-induced HF have been used extensively to show the benefit of recombinant neuroglin-1β (Galindo et al., 2014). Treatment with neuroglin-1β for 7 days was sufficient to prevent ventricular remodeling following volume-overload induced cardiac hypertrophy (Wang et al., 2019). At the molecular level, neuroglin-1β partially prevented the decrease in SERCA2a, LTCC and increase in NCX1 associated with HF (Wang et al., 2019). Therefore, neuroglin-1β may confer its cardioprotective effects through regulation of Ca2+-handling protein expression. Given the promising effects of recombinant neuroglin-1β treatment in animal models of HF, Phase II and III clinical trials were approved to determine the safety and efficacy of daily infusions of Neucardin (Galindo et al., 2014). The first Phase II clinical trials successfully demonstrated the safety, tolerability and efficacy of rhNRG-1 in patients with chronic HF (Gao et al., 2010; Jabbour et al., 2011) [clinicaltrials.gov identifier NCT01251406 and NCT01541202]. Following on from these early Phase II clinical trials, large-scale, multi-centre Phase III clinical trials have begun recruiting patients to determine the effect of Neucardin on all-cause mortality after 1 year [clinicaltrials.gov NCT03388593]. Furthermore, a Phase I clinical trial has recently been accepted to test the safety, tolerability and immunogenicity of the drug JK07 in HFrEF patients; JK07 is a human immunoglobulin IG1 monoclonal antibody containing an active polypeptide fragment of neuroglin-1 [clinicaltrials.gov identifier NCT04210375] (Figure 4 and Table 2). Future studies should aim to determine whether neuroglin-1β mediates NCX1, SERCA2a and LTCC channel expression in other animal models of HF and whether the efficacious effects of Neucardin in clinical trials is partly due to altered Ca2+ dynamics.
RyR2
There is a general consensus that SR-located RyR2 contributes to Ca2+ leak during HF which can cause (1) cardiac arrhythmias, (2) reduced Ca2+ SR content, (3) systolic dysfunction, and (4) altered cardiac energetics (Bers, 2014). The function of RyR2, the major cardiac isoform of the RyR, is tightly regulated though changes to its phosphorylation status, Ca2+ activation and inactivation sites and post-translational modifications (Roe et al., 2015). RyR2 channel gating is regulated by accessory proteins whilst its tetrameric confirmation is stabilized by FK506-binding protein-12.6 (FKBP12.6). Disassociation of FKBP12.6 from RyR2 is observed in some models of HF and is achieved through PKA-mediated hyperphosphorylation of RyR2 at Ser2808 (Marx et al., 2000). One study has demonstrated that transgenic mice with an absent PKA phosphorylation site on RyR2 are protected against MI-induced HF (Wehrens et al., 2006); however, others have shown that CaMKII-mediated RyR2 phosphorylation leads to altered Ca2+ sparks as opposed to PKA-mediated phosphorylation (Li et al., 2002; Guo et al., 2006). Such is the importance of CaMKII-mediated phosphorylation of RyR2 that it has been shown to promote the transition from hypertrophy into HF (Ling et al., 2009; Respress et al., 2012).
Changes to Ca2+ transfer between the SR and mitochondria impedes mitochondrial function in cardiomyocytes (Fernandez-Sanz et al., 2014; Ruiz-Meana et al., 2019), potentially contributing to cardiomyocyte deterioration in HF. Recently, a novel mechanism of RyR2 hyperglycation was shown to increase Ca2+ leak from the SR in cardiomyocytes of aged mice (Ruiz-Meana et al., 2019). Aging alone results in advanced glycation of RyR2, leading to increased SR Ca2+ leak, mitochondrial Ca2+ overload and mitochondrial dysfunction, independently of cardiac disease (Ruiz-Meana et al., 2019). Importantly, hyperglycation of RyR2 and increased mitochondrial Ca2+ was prevalent in atrial appendages from aged patients (75 years or above) and absent in young patients (Ruiz-Meana et al., 2019). Preventing the glycation of RyR2 by inhibiting dicarbonyl intermediaries or stimulating their detoxification may provide a preventative measure to attenuate mitochondrial damage associated with aged, failing cardiomyocytes.
RyR2 can be inhibited by dantrolene sodium, a derivative of the hydantoin class of compounds (Maxwell et al., 2012). Application of dantrolene normalizes the diastolic Ca2+ leak seen in HF; however, owing to its hepatotoxic side-effect, dantrolene is not suitable for long-term use (Durham et al., 1984) (Figure 4 and Table 2). Despite this, the ‘SHO-IN’ clinical trial seeks to determine the safety and efficacy of dantrolene on patients with chronic HF [Japanese Registry of Clinical Trials identifier 61180059] (Kobayashi et al., 2020). Recent work has shown that phenytoin, a member of the same class of hydantoin compounds, shares the same RyR2 inhibitory effects as dantrolene (Ashna et al., 2020). In vitro application of phenytoin to RyR2 channels isolated from failing human hearts shows that phenytoin reversibly inhibits RyR2 which subsequently inhibits diastolic cytoplasmic, but not systolic, Ca2+ release (Ashna et al., 2020). Interestingly, phenytoin is currently used clinically as a Na+ channel blocker to treat epilepsy, therefore there is scope to determine the potential therapeutic use of phenytoin in HF (Figure 4). Designing novel therapeutic targets for RyR2 may, in future, involve RyR2 accessory proteins which modulate its function. TRIC-A and B are SR-located K+ channels known to participate in ECC (Yazawa et al., 2007; Yamazaki et al., 2009b; Yang et al., 2016); both TRIC-A and TRIC-B knockout mice exhibit Ca2+ overload in the SR (Yamazaki et al., 2009a; Zhao et al., 2010). TRIC-A, but not TRIC-B, physically associates with RyR2 to enhance RyR2-dependent Ca2+ efflux from the SR; however, there is still a limited understanding of the function of TRIC-A-dependent RyR2 modulation under pathological conditions (Zhou et al., 2020).
TRP Channels
When compared to a healthy myocardium, TRPC1, C5, M4, and M7 are upregulated and TRPC4 and V2 are downregulated in the failing human heart (Dragun et al., 2019), suggesting each TRP channel may differentially contribute to CVD [reviewed extensively elsewhere (Falcon et al., 2019; Hof et al., 2019)]. Interestingly, in neonatal rat cardiomyocytes, stimulation with hypertrophy-inducing agents leads to the upregulation and subsequent activation of TRPC1 (Ohba et al., 2007) and C5 (Sunggip et al., 2018). Furthermore, in mouse models, global deletion of Trpc1 attenuates pressure-overload induced hypertrophy through reduced activation of the calcineurin/NFAT pathway (Ohba et al., 2007; Seth et al., 2009). In addition to TRPC1 and C5, TRPC3 (Brenner and Dolmetsch, 2007) and C7 (Satoh et al., 2007) are also upregulated in hypertrophy-stimulated cardiomyocytes and TRPC6 overexpression induces cardiac hypertrophy in mice (Kuwahara et al., 2006). TRPC3 and C6 have been successfully co-targeted with the thiazole inhibitors GSK2332255B (GSK255B) and GSK2833503A (GSK503A), respectively (Washburn et al., 2013) (Figure 4 and Table 2). When these inhibitors were co-administered following pressure-overload induced hypertrophy in mice, both cardiomyocyte hypertrophy and cardiac fibrosis were reduced (Seo et al., 2014). Mechanistically, blockade of TRPC3 reduces phosphorylation of Ca2+-handling proteins in mouse embryonic stem cell-derived cardiomyocytes (Qi et al., 2016), which may alter downstream Ca2+-mediated signaling. More specifically, TRPC3 can be exclusively inhibited by Pyr-3 (Kiyonaka et al., 2009) (Figure 4 and Table 2). Pyr-3 inhibition of TRPC3 attenuates NFAT-mediated transcription and can prevent hypertrophy in vitro in ANG-II stimulated neonatal rat cardiomyocytes and in vivo following pressure overload-induced hypertrophy in mice (Kiyonaka et al., 2009). Other studies have also shown TRPC1 and TRPC3-C6 upregulation in failing human hearts when compared to non-failing myocardium (Bush et al., 2006; Morine et al., 2016). Interestingly, the same TRPC channels are also upregulated in the border zone of rats hearts after left anterior descending coronary artery ligation-induced MI (Dominguez-Rodriguez et al., 2018). Therefore, it appears that the TRPC family of channels contribute to the cardiac HF phenotype; however, more research is required in order to determine the functional effect of each upregulated TRPC channel in order to progress pre-clinical studies.
The TRPV family have also been extensively investigated; however, as mentioned above, there is no detectable upregulation of TRPV family members in the failing human myocardium (Dragun et al., 2019). Despite this, there is promising data to suggest blockade of TRPV4 may prove beneficial in HF patients; the pulmonary endothelial-localized TRPV4 channel has been shown to play a role in pulmonary venous pressure-induced formation of pulmonary oedema in HF (Thorneloe et al., 2012). In support of this, TRPV4 is upregulated in human lungs derived from patients with congestive HF (Thorneloe et al., 2012). As such, TRPV4 channel blockade with the first in class orally active pharmaceutic GSK2798745 successfully prevents the formation of and reverses established pulmonary oedema in acute and chronic HF models (Thorneloe et al., 2012) (Figure 4 and Table 2). The positive data obtained from TRPV4 inhibition in animal models led to a Phase I clinical trial to investigate the safety and efficacy of inhibiting TRPV4 with GSK2798745 (Goyal et al., 2019). Results of this first in man trial has shown there are no adverse effects or safety issues in both healthy volunteers and patients with mild to moderate HF (Goyal et al., 2019) [clinicaltrials.gov identifiers NCT02497937 and NCT02119260]. In addition, TRPV4 is activated in the murine model of I/R injury (Dong et al., 2017). Mechanistically, activation of TRPV4 following I/R leads to increased Ca2+ influx and generation of ROS (Wu et al., 2017). Therefore, chemical inhibition of TRPV4 with the selective antagonist HC-067047 prevents aberrant TRPV4 activation which leads to a reduction in infarct size and improved cardiac function following I/R injury (Dong et al., 2017). Furthermore, both TRPV1 and V2 contribute to the inflammatory response following MI; however, mice harboring a global deletion of TRPV1 show an exacerbated inflammatory response 3- and 7-days post-MI whereas mice with a global knockout of TRPV2 show improved cardiac recovery following MI due to a reduction in peri-infarct macrophages (Huang et al., 2009; Entin-Meer et al., 2017; Falcon et al., 2019).
Some of the TRPM and TRPP channels exhibit the highest levels of expression in basal ventricular myocytes, amongst these are TRPM7, M1 and M4 and TRPP1 and P2 (Chevalier et al., 2018). As mentioned, both TRPM4 and M7 levels are elevated in the failing human ventricle myocardium (Dragun et al., 2019); however, TRPM7 mRNA was shown to be reduced in the left ventricle of patients with ischemic cardiomyopathy (Ortega et al., 2016), therefore the underlying cause of HF may influence the expression pattern of TRP channels. TRPM7 is expressed in the SAN and has a known role in modulation of cardiac automaticity (Sah et al., 2013a,b); however, TRMP7 also promotes cardiomyocyte proliferation during early cardiogenesis (Sah et al., 2013a,b). TRMP7 kinase dead mice exhibit significant cardiac hypertrophy and upregulation of pro-inflammatory mediators, driven by a Mg2+- but not a Ca2+-sensitive mechanism (Rios et al., 2020). Therefore, TRPM4 may provide a better option to therapeutically target Ca2+ mishandling. Interestingly, despite the upregulation of TRPM4 in failing human hearts, the channel itself is impermeable to Ca2+; however, it is both Ca2+- and voltage-activated (Nilius et al., 2003, 2005; Wang et al., 2018). TRMP4 expression is upregulated by 50-fold in spontaneously hypertensive rats when compared to controls (Guinamard et al., 2006). Furthermore, global deletion of Trpm4 causes eccentric cardiac hypertrophy with aging which is caused by neonatal cardiomyocyte hyperplasia (Demion et al., 2014). In a cardiomyocyte-specific Trpm4 knockout mouse (Trpmcko), induction of cardiac hypertrophy with ANG-II resulted in increased hypertrophy in Trpm4cko mice when compared to controls (Kecskes et al., 2015). TRPM4 activity can be exclusively inhibited by 9-phenanthrol (Guinamard et al., 2014). Most studies using 9-phenanthrol have aimed to determine the contribution of TRMP4 to smooth muscle cell contraction (Guinamard et al., 2014); however, application of 9-phenanthrol to isolated rat hearts in a model of I/R injury reduces infarct size and improves contractility (Wang J. et al., 2013).
PMCAs
The PMCAs are rapidly emerging as potential signaling mediators (Armesilla et al., 2004; Oceandy et al., 2007; Cartwright et al., 2011; Stafford et al., 2017); however, to date, only a role for PMCA4 has been elucidated in a mouse model of TAC-induced pressure-overload hypertrophy. In the study by Mohamed et al. (2016), global knockout of Atp2b4 was shown to attenuate the pressure-overload response; however, the anti-hypertrophic effects of PMCA4 knockout could only be replicated in fibroblast-specific and not in cardiomyocyte-specific knockout mouse models (Mohamed et al., 2016; Stafford et al., 2017). Non-myocyte cells such as cardiac fibroblasts and endothelial cells account for around 70% of total cardiac cells (Pinto et al., 2016), and are able to secrete factors which can influence adjacent cardiomyocyte signaling. Interestingly, fibroblast-specific deletion of PMCA4 leads to significant upregulation of secreted frizzled related protein 2 which is thought to confer cardioprotective properties by inhibiting the Wnt pathway in neighboring cells (Mohamed et al., 2016). Despite this role for PMCA4 in cardiac hypertrophy, there is no evidence that human ATP2B4 is associated with CVD; genome-wide association studies (GWAS) have to date been unable to associate SNPs within ATP2B4 with any CVD (Tabara et al., 2010).
In contrast to GWAS data for ATP2B4, SNPs located in ATP2B1, the gene encoding PMCA1, have been strongly associated with CVDs, including hypertension, coronary artery disease and MI (Tabara et al., 2010; Takeuchi et al., 2010; Wang Y. et al., 2013; Xu et al., 2016; Jamshidi et al., 2018; Sombie et al., 2019). The association of ATP2B1 with hypertension has been shown in numerous studies across different ethnic groups; to date, there are at least six different SNPs within ATP2B1 which are associated with elevated blood pressure (Tabara et al., 2010; Takeuchi et al., 2010; Wang Y. et al., 2013; Xu et al., 2016; Jamshidi et al., 2018; Sombie et al., 2019). In mice, vascular smooth muscle cell-specific PMCA1 knockout mice exhibit elevated blood pressure (Kobayashi et al., 2012). Mechanistically, loss of PMCA1 from vascular smooth muscle cells leads to enhanced expression of LTCC and increased Ca2+ concentration which may affect smooth muscle cell stiffness and subsequently, vascular tone (Okuyama et al., 2018; Zhu et al., 2019). Furthermore, PMCA1 global heterozygous mice have increased hypertension with aging, which is preceded by vascular remodeling (Little et al., 2017). Taken together, PMCA1 appears to play a functional role in the development of hypertension through modulation of Ca2+. Whilst more research is required to determine the contribution of PMCA1 to hypertrophy and HF, there is scope to therapeutically target PMCA1 as a modulator of hypertension to prevent CVD (Little et al., 2016).
The PMCAs are typical ATPases and therefore present a suitable novel therapeutic target (Figure 4 and Table 2). ATPases have been clinically targeted to treat hypotension and some cardiac arrhythmias; ouabain inhibits Na+/K+-ATPase and omeprazole inhibits H+/K+-ATPase. Therapeutically, the two main cardiac isoforms of PMCA have been successfully targeted in vitro; the caloxins, a class of PMCA inhibitors, show differential specificity for each PMCA isoform (Pande et al., 2011). Caloxin 1B3 shows increased specificity for PMCA1 over PMCA4 and results in increased Ca2+ in endothelial cells (Szewczyk et al., 2010; Pande et al., 2011), whereas caloxin 1B1 is predominantly PMCA4-isoform specific (Pande et al., 2006). PMCA4 has also been successfully targeted with aurintricarboxylic acid (Mohamed et al., 2013a). Application of aurintricarboxylic acid in vivo was able to prevent and also reverse hypertrophy in the murine TAC model of pressure overload (Mohamed et al., 2013b), suggesting inhibition of PMCA4 may provide an interesting therapeutic target.
MCU
Ca2+ is also required for effective mitochondrial function. Mitochondrial Ca2+ cycling is achieved through the inner mitochondrial membrane located MCU (Figure 2). Mitochondria are abundant in cardiomyocytes owing to their function in the synthesis of ATP (Kwong, 2017). In the process of ATP generation, mitochondria utilize Ca2+ to activate pyruvate dehydrogenase, ATP synthase and other enzymes involved in the redox reaction, as such [Ca2+] in the inner matrix is tightly regulated by MCU (Kwong, 2017). MCU levels are elevated in the failing human heart and in animal models of HF (Zaglia et al., 2017; Yu et al., 2018). Under pathological conditions, overload of mitochondrial Ca2+ is associated with reduced cell viability, as well as apoptotic and autophagic cell death (Pinton et al., 2008). Interestingly, recent work suggests MCU is dispensable for normal cardiac function; cardiomyocyte-specific Mcu–/– mice show no overt phenotype under basal conditions, suggesting MCU is not required for maintaining cardiac homeostasis (Luongo et al., 2015). Furthermore, Mcu–/– mice do not experience mitochondrial Ca2+ overload following I/R injury which protects them against cell death and adverse cardiac remodeling (Luongo et al., 2015). Similarly, following TAC-induced pathological cardiac hypertrophy, mice treated with the MCU chemical inhibitor RR show improved survival, cardiac function and maintained mitochondrial integrity (Yu et al., 2018) (Figure 4 and Table 2). Treatment of isoprenaline stressed H9C2 cells with RR in vitro confirmed its ability to increase both autophagy and mitophagy (Yu et al., 2018). Taken together, inhibition or depletion of MCU appears beneficial for attenuating the pathological response to ischemic and hypertrophy stimuli. However, the use of RR has been shown to interfere with Ca2+ flux through mitochondrial-located RyR1 and membrane bound LTCC (Beutner et al., 2001). Therefore, developing more specific inhibitors of MCU to combat HF offers an alternative to targeting the ‘typical’ Ca2+-handling proteins.
Cardiac Arrhythmias
Cardiac arrhythmias are a class of conditions describing abnormalities in the heart’s normal rhythm. Arrhythmias are a leading cause of morbidity and mortality, and can lead to stroke, deterioration of ventricular function, and sudden cardiac death (Kass and Clancy, 2006). Arrhythmic events can vary in terms of speed, rhythm, duration and location in the heart, and can present in numerous ways. The main types of arrhythmia include bradycardias (slow but regular heart rhythms) and tachycardias (faster than normal heart rates), including supraventricular tachycardias (tachycardia originating in the atria). Other common arrhythmias are AF (an irregular, often fast heart rhythm) and heart block (the heart beats slower than normal due to a delay in conduction) (Keating and Sanguinetti, 2001; Kass and Clancy, 2006).
Arrhythmogenesis is complex and multifactorial. Numerous genetic mutations have been shown underlie primary arrhythmia disorders, such as LQTS and CPVT. Within these conditions, the underlying genetic defect determines the arrhythmia phenotype, including the physiological triggers which incite arrhythmic events in these patients (Wilde and Bezzina, 2005). In addition, pathological and physiological factors can influence cardiac electrical remodeling, and therefore trigger arrhythmia development, including HF, hypertension, and coronary heart disease. For example, HF can result in structural and hemodynamic dysfunction, electrical remodeling (including those impacting Ca2+ handling), and metabolic changes which pre-dispose the development of arrhythmias (Masarone et al., 2017). Furthermore, susceptibility to these acquired arrhythmic disorders can be influenced by genetic factors (Wilde and Bezzina, 2005).
Calcium Handling Proteins in Arrhythmia Development and Their Potential as Novel Therapeutic Targets
Calcium handling in the myocardium is key to the generation and control of the heartbeat. Intracellular Ca2+ remodeling has been identified as a major factor in the development of arrhythmias and current approved therapies for arrhythmic disorders include Ca2+ channel blockers (Deo et al., 2017). Advancements in human genetic studies have highlighted mutations in Ca2+ handling proteins, as well as Ca2+ channels, that can result in the development of arrhythmias. In addition, remodeling of Ca2+ handling proteins has been identified in patients with acquired arrhythmic conditions (Landstrom et al., 2017). Therefore, Ca2+ handling proteins are promising therapeutic targets for the treatment of arrhythmic disorders. In the following section, we will discuss the current research aimed at targeting Ca2+ handling proteins in arrhythmia development.
The Role of Ca2+ in Arrhythmia Development
The mechanisms underlying arrhythmia development are generally divided into three categories: automaticity, triggered activity, and re-entry (Antzelevitch and Burashnikov, 2011). The role of Ca2+ in the generation of arrhythmias has been previously extensively reviewed (see Antzelevitch and Burashnikov, 2011; Kistamas et al., 2020). Automaticity is the ability of a cell to spontaneously depolarize and generate an AP. Under normal conditions, automaticity is restricted to the cardiac conduction system, specifically the SAN pacemaker cells which initiate cardiac contraction (Antzelevitch and Burashnikov, 2011). Abnormal automaticity occurs when the automaticity of the SAN is altered or when other cells begin to fire spontaneously. Spontaneous beating of the SAN is regulated by a coupled system of the surface membrane clock (inactivation and deactivation of cardiac ion channels) and the intracellular Ca2+ clock (rhythmic SR Ca2+ release) (Joung et al., 2011). Unsurprisingly, malfunction of both clocks has been associated with arrhythmia development. In relation to Ca2+ handling, Ca2+ clock remodeling (unresponsiveness to β-adrenergic stimulation and downregulation of RyR2) has been identified in SAN dysfunction (Joung et al., 2011). In addition, triggered oxidation and activation of CaMKII has been shown to result in SAN cell death which may contribute to SAN dysfunction (Huke and Knollmann, 2011).
Triggered activity refers to voltage oscillations termed afterdepolarizations which are often Ca2+-mediated. Early afterdepolarizations (EADs) occur during the plateau phase of the cardiac AP and are associated with LQTS and ventricular tachyarrhythmias (associated with HF) (Landstrom et al., 2017). They are triggered by a reduction in repolarization reserve as a result of an increase in an inward current or a decrease in an outward current (Huang et al., 2018). A main contributor to EAD initiation is an increase in L-type Ca2+ current (ICaL) which drives the EAD upstroke. This can occur due to a reactivation of the current during the repolarization phase caused by a reduction in the outward IK current or by CaMKII-mediated phosphorylation of ICaL (Weiss et al., 2010; Landstrom et al., 2017). In addition, increased NCX current (INCX) can contribute to EAD. Spontaneous Ca2+ release events either prior to the completion of repolarization or during the initial phase of EADs can cause an increase in INCX activity which may delay repolarization and allow ICaL to reactivate (Zhong et al., 2018).
Delayed afterdepolarizations (DADs) typically occur after repolarization and are caused by Ca2+ overload driving spontaneous Ca2+ release from the SR (Fink et al., 2011). Ca2+ release from the SR activates NCX (Ca2+-sensitive Cl–- and non-specific ion currents can also be activated) leading to depolarization of the membrane. If the excitation threshold is reached, INa becomes activated and an AP is triggered (Fink et al., 2011). An increase in Ca2+ influx or a decrease in Ca2+ efflux can lead to an increase in SR Ca2+ content and drive the Ca2+ waves that activate NCX. The SR threshold for Ca2+ waves is thought to be dependent on the properties of RyR, with the opening of RyR decreasing the threshold for Ca2+ release (Venetucci et al., 2008). Elevated Ca2+ levels increase the open probability of RyR, making them more sensitive to stochastic Ca2+ release, ultimately leading to Ca2+ release (Fink et al., 2011). Phosphorylation of RyR at the PKA site or the CaMKII site can also increase RyR calcium sensitivity and open probability (Wehrens et al., 2004a). Modulation of RyR has been attributed to arrhythmias associated with HF and CPVT (caused by an underlying RyR mutation) (Venetucci et al., 2008).
Re-entry underlies the formation of arrhythmias due to abnormal impulse conduction. Here an electrical impulse fails to terminate and instead re-excites the myocardium. For re-entry to occur there must be an abnormal electrical circuit, slow conduction and a unidirectional block (Antzelevitch and Burashnikov, 2011). Changes to Ca2+ handling can cause cardiac tissue to become vulnerable to re-entry arrhythmias by increasing the dispersion of refractoriness, leading to unidirectional conduction block, and slowing of conduction (Weiss et al., 2011). Subthreshold EADs and DADs cause electrical homogeneity by increasing dispersion of refractoriness and dispersion of excitability, respectively (Landstrom et al., 2017; Kistamas et al., 2020). In addition, cardiac alternans can act as a substrate from the initiation of re-entry and can arise due to Ca2+ cycling dysfunction. Cardiac alternans present as beat to beat variations in Ca2+ transient amplitude (Ca2+ alternans), AP duration (APD alternans), or contraction amplitude (mechanical alternans) (Kistamas et al., 2020). Changes in the level of SR Ca2+ content, reduced SERCA activity, and changes in the refractoriness have been proposed as mechanisms for the generation of Ca2+-derived alternans. DADs can also result in Ca2+ alternans and subsequent APD alternans (Kistamas et al., 2020).
Ca2+-Handling Proteins in Arrhythmia Development; Ca2+ Channels, Pumps and Exchangers
RyR2
Abnormal RyR2-mediated Ca2+ release from the SR can lead to both atrial and ventricular arrhythmias, some primary arrhythmia disorders including CPVT and forms of acquired arrhythmias associated with heart disease (AF during HF) (McCauley and Wehrens, 2011). To diminish arrhythmic events, therapies have been developed to reduce and stabilize abnormal RyR activity, in order to inhibit diastolic SR Ca2+ release whilst maintaining the normal SR Ca2+ release during systole.
Dantrolene sodium is an approved muscle relaxant which is used for the treatment of malignant hyperthermia, caused by mutations in the skeletal RyR (RyR1) (McCauley and Wehrens, 2011). Dantrolene binds to the N-terminal of RyR1 to stabilize the inter-domain interactions required for the closed state of the RyR Ca2+ channel (Kobayashi et al., 2005). Further studies have shown dantrolene binds to RyR2 in a similar manner to improve the function of the receptor, suggesting the use of dantrolene in the treatment of arrhythmias (Figure 4) (Kobayashi et al., 2009). In a study using a knock-in mouse model carrying a human CPVT-RyR2 mutation (R2475S), dantrolene inhibited CPVT-related tachycardia (Kobayashi et al., 2010). Furthermore, in a cohort of CPVT-1 patients, dantrolene had an anti-arrhythmic effect, and reduced the number of premature ventricular complexes. This anti-arrhythmic response was also evident in induced pluripotent stem cell derived CPVT1 cardiomyocytes (Penttinen et al., 2015). In addition to CPVT, dantrolene has been shown to prevent the occurrence of DADs in failing cardiomyocytes isolated from a canine model (Kobayashi et al., 2009) and have a protective effect against arrhythmic events in animal models of I/R (Balam Ortiz et al., 1999; Boys et al., 2010). Together the data suggest dantrolene is a promising therapy to attenuate Ca2+-mediated arrhythmias associated with HF. An early Phase I clinical trial aimed to assess the feasibility of intravenous delivery of dantrolene to reduce ventricular arrhythmias in patients with structural heart disease [clinicaltrials.gov identifier NCT04134845]. While no results have yet been posted, the study highlights the potential of RyR2 inhibition for the treatment of arrhythmias.
JTV519 (also known as K201) is a 1,4-Benzothiazepines derivative initially identified to suppress sudden cardiac cell death associated with Ca2+ overload (Kaneko et al., 2009). Further studies identified RyR2 as a target of JTV519, with JTV519 having a protective effect in models of AF as a result of the restoration of RyR function (Figure 4 and Table 2) (Kumagai et al., 2003; Vest et al., 2005). PKA hyperphosphorylation of RyR2 and a reduction in the RyR2 channel-stabilizing subunit calstabin2 has been identified in AF. In a canine model of AF, treatment with JTV519 restored calstabin2 levels, stabilizing the closed state of RyR2 and reducing Ca2+ leak (Vest et al., 2005). A Phase 2 clinical trial evaluated the effects of JTV519 on sinus rhythm restoration in patients with AF [clinicaltrials.gov identifier NCT00626652], although no data is available. In terms of ventricular arrhythmias, a study using a calstabin2 global heterozygous knockout found pre-treatment with JTV519 prevented exercise-induced ventricular arrhythmias and sudden cardiac death associated with depletion of calstabin2. In the same study, JTV519 had no effect on calstabin2 knockout mice highlighting the requirement of calstabin2 for the antiarrhythmic effect of JTV519 (Wehrens et al., 2004b). Another 1,4-Benzothiazepines derivative is S107 which, like JTV519, can enhance RyR2 and calstabin2 binding to stabilize RyR2. S107 has been shown to be more specific than JTV519, with the latter known to interact with other cardiac ion channels including LTCC (Andersson and Marks, 2010). In a mouse model, S107 increased the binding of calstabin2 to RyRs carrying a CPVT mutation (R2474S) and inhibited associated arrhythmias (Figure 4) (Lehnart et al., 2008). The antiarrhythmic effect of S107 was further identified using a human induced pluripotent stem cell model of CPVT (Sasaki et al., 2016).
Flecainide is a trifluoroethoxybenzamide approved for the treatment of AF and/or supraventricular tachycardia. Importantly, flecainide is not recommended to patients with underling structural heart disease as the drug can be pro-arrhythmic in this environment (Andrikopoulos et al., 2015). While flecainide is classed as a type IC antiarrhythmic drug (a Na+ channel blocker) it is worth noting here the drugs ability to inhibit RyR2 action by reducing the duration of channel opening (Figure 4). Through this role as an RyR inhibitor, flecainide has been shown, in a mouse model of CPVT, to prevent ventricular tachycardia by suppressing spontaneous Ca2+ release. In addition, flecainide prevented exercise-induced ventricular arrhythmias in two patients carrying a CPVT-linked RyR mutation who were refectory to conventional therapy (Watanabe et al., 2009).
NCX
NCX has been identified to have a dual role in arrhythmogenesis. When Na+ levels are high, NCX acts in the reverse model to bring Ca2+ into the cell which can result in Ca2+ overload. When acting in the forward mode, the inward current formed during Ca2+ release can result in DADs (Antoons and Sipido, 2008). Increased NCX activity has been shown to underlie arrhythmogenesis in numerous conditions including HF and AF, making NCX inhibition an attractive arrhythmia treatment.
First generation NCX blockers such as aprinidine and bepridil displayed anti-arrhythmic effects. However, these NCX blockers lacked selectivity and were suggested to only have modest NCX blocking effects at therapeutic concentrations (Hobai and O’Rourke, 2004). KB-R7943 was the first NCX blocker with reported mode-selectivity, shown to be a more specific blocker of NCX in the reverse mode (Elias et al., 2001). Animal studies have shown KB-R7943 to be protective against Ca2+ overload and able to suppress reoxygenation-induced arrhythmias (Figure 4) (Ladilov et al., 1999; Mukai et al., 2000). In addition, KB-R7943 suppressed afterdepolarizations and ventricular tachyarrhythmias in a rabbit MI mode l (Chang et al., 2015). Paradoxically, this study also found KB-R7943 to enhance the inducibility of arrhythmias in this model (Chang et al., 2015), with another study showing KB-R7943 had no effect on I/R arrhythmias (Miyamoto et al., 2002). Further studies have also shown KB-R7943 to inhibit the LTCC current and various K+ currents, which brings into question the use of the drug in the treatment of arrhythmias (Tanaka et al., 2002).
SEA-0400 has been described as the most potent and selective NCX inhibitor compared to KB-R7943 (Tanaka et al., 2002). Like KB-R7943, SEA-0400 has been suggested to have a higher potency for the forward NCX mode (Lee et al., 2004) and has been shown to have a cardioprotective and antiarrhythmic effect, although results vary in different models (Figure 4). In a rat model of myocardial I/R injury, SEA-0400 improved cardiac function, and reduced the incidence of ventricular fibrillation and mortality (Takahashi et al., 2003). In contrast, however, the drug had no effect on the incidence of ventricular tachycardia and fibrillation in a larger animal model of I/R injury (Nagasawa et al., 2005). This study also investigated arrhythmias induced by digitalis. Here, SEA-0400 reduced the incidence of induced tachyarrhythmias although 2/8 animals experienced AV block and cardiac standstill (Nagasawa et al., 2005). This differential arrhythmic effect may be dependent on the expression levels of NCX. A study found SEA-0400 to be antiarrhythmic in mouse models with increased NCX expression, such as HF and AF, but in conditions with reduced NCX expression, SEA-0400 was found to increase Ca2+ transient amplitude and spontaneous Ca2+ transients, ultimately acting proarrhythmic (Bogeholz et al., 2017). These findings suggest that in clinical conditions in which NCX function is reduced, such as diabetic cardiomyopathy, further NCX inhibition could trigger arrhythmias by reducing NCX function below the level required for balanced Ca2+ dynamics (Bogeholz et al., 2017).
The antiarrhythmic benefits of blocking NCX with KB-R7943 and SEA-0700 has driven further studies to identify novel NCX inhibitors including SN-6 and ORM-10962 (Figure 4 and Table 2). While SN-6 has been shown to have cardioprotective effects, the potential of the benzyloxyphenyl derivative in treating HF-associated arrhythmias is hindered by its ability to also increase diastolic Ca2+ and block ICa (Gandhi et al., 2013). ORM-10962 is a potent NCX blocker which can inhibit both forward and reverse NCX currents and which has no significant effect on other cardiac ion currents. The drug has been shown to attenuate digoxin-induced DAD in isolated ventricular Purkinje fibers but had no effect on arrhythmias related to I/R injury (Kohajda et al., 2016). The contrasting effects of NCX blockers on arrhythmogenesis under different conditions highlights the need for further studies to explore the use of NCX blockers.
Interestingly, another therapeutic approach has been to stimulate the forward mode of NCX to reduce cytosolic Ca2+ and attenuate Ca2+ overload. Heparin oligosaccharides have been shown to directly interact with NCX and accelerate its activity (de Godoy et al., 2018). In a model of I/R, trisulfated heparin disaccharide and low molecular weight heparins had a significant antiarrhythmic effect, as a result of increased NCX forward activity. While it is worth nothing that both agents are known to target LTCC and Na+ channels, the mechanism of NCX acceleration appears to be a promising antiarrhythmic target (de Godoy et al., 2018).
SERCA
SERCA and its inhibitory protein PLN have been identified as important regulators of cardiac contractility and therefore potential antiarrhythmic targets (Antoons and Sipido, 2008). Impaired SERCA activity in HF results in dysregulation of systolic and diastolic function which can lead to cardiac arrhythmias. Increasing SERCA activity is a way of improving cardiac function in HF. In terms of arrhythmogenesis an increase in SERCA activity could theoretically result in Ca2+ overload and ultimately be arrhythmogenic (Antoons and Sipido, 2008). Encouragingly however, studies focusing on HF have found an increase in SERCA2 expression to be antiarrhythmic. In pre-clinical models of HF, increased SERCA2 expression through adenoviral delivery (Ad.SERCA2a) improved intracellular Ca2+ handling, restored contractility and attenuated ventricular arrhythmias (Figure 4) (del Monte et al., 2004; Lyon et al., 2011; Cutler et al., 2012). Other direct therapies to activate SERCA2 or inhibit PLN binding have been studied. Istaroxime, a Na+/K+ ATPase inhibitor which also stimulates SERCA, has been shown to have a moderate antiarrhythmic effect (Figure 4) (Bossu et al., 2018). Several clinical trials have assessed istaroxime in HF, highlighting the promise of the drug in cardiac disorders. However, it is worth noting is not known if higher doses of istaroxime could stimulate further arrhythmic episodes through Na+/K+ ATPase inhibition (Bossu et al., 2018).
T-Type Ca2+ Channel
As previously described, LTCC blockers are an approved treatment strategy for some arrhythmic conditions. The T-type Ca2+ channel has been also investigated as an antiarrhythmic target. T-type Ca2+ channels are localized in the pacemaker regions of the heart (the SAN, AVN and Purkinje fibers) where they are thought to contribute to SAN pacemaker activity and atrioventricular conduction (Mangoni et al., 2006). Inactivation of T-type Ca2+ channels results in bradycardia and delayed atrioventricular conduction, highlighting the importance of the channel in cardiac rhythm (Mangoni et al., 2006). Several studies have investigated the effect of blocking T-type Ca2+ channels with mibefradil, a tetralol derivative which inhibits T-type Ca2+ channels but is also known to block ICaL. Treatment with mibefradil results in a dose-dependent decrease in heart rate (Brogden and Markham, 1997), and prevention of ischemic-induced ventricular fibrillation (Figure 4) (Billman and Hamlin, 1996). However, the drug has been reported to result in toxicity, inducing TdP due to QTc prolongation (Glaser et al., 2001).
CaMKII
CaMKII dysfunction is a common hallmark of many cardiac pathologies including HF. CaMKII-mediated arrhythmias have been identified in HF, AF, reperfusion injury, and genetic arrhythmic conditions including LQTS. Changes in CaMKII expression can influence the function of key cardiac ion channels and result in arrhythmogenesis (Mustroph et al., 2017). CaMKII-mediated hyperphosphorylation of RyR2 leads to spontaneous Ca2+ release events which can be proarrhythmic in disease conditions associated with Ca2+ leak. CaMKII can also drive Ca2+ overload through phosphorylation of Nav1.5 and subsequent reduction in NCX-mediated Ca2+ extrusion. In addition, an increase in CaMKII activity can result in prolonged repolarization through an increase in ICaL, an increase in Na+ influx, and reduced IK function (Mustroph et al., 2017). This makes the inhibition of CaMKII is a promising antiarrhythmic approach.
KN-93, a CaMKII inhibitor, which works by binding directly to Ca2+/CaM, disrupts the ability of Ca2+/CaM to interact with CaMKII and activate the kinase (Wong et al., 2019). Inhibition of CaMKII by KN-93 has been shown to be antiarrhythmic in several models (Figure 3). In a RyR2R4496C± knock-in mouse model of CPVT, KN-93 prevented catecholamine-induced sustained ventricular tachyarrhythmias (Liu et al., 2011). Further studies using induced pluripotent stem cells generated from a CPVT patient carrying a RyR2 mutation, showed KN-93 to drastically reduce DADs induced by catecholaminergic stress as well as stabilize Ca2+ activation under β-adrenergic stimulation to a pattern seen in normal tissue (Di Pasquale et al., 2013). CaMKII inhibition by KN-93 was also shown to reduce the incidence of stress-induced ventricular tachycardia in a junction knock-out mouse model which presents with a phenotype similar to CPVT (RyR2 channels display CaMKII-dependent hyperphosphorylation). Here, KN-93 also reduced the number of spontaneous Ca2+ aftertransients and aftercontractions under stress conditions (Tzimas et al., 2015). It is worth noting that in relation to CPVT, AAV-mediated delivery of a CaMKII peptide inhibitor (autocamtide-2-related inhibitory peptide, AIP) has been shown to suppress ventricular arrhythmias induced by either β-adrenergic stimulation or programmed ventricular pacing. AAV-GFP-AIP delivery also suppressed abnormal Ca2+ release events in induced pluripotent stem cells derived from two patients with two distinct CPVT pathogenic mutations (Bezzerides et al., 2019). The advantage of using a gene therapy approach over classic CaMKII blockers, including KN-93, is that it allows for cardiomyocyte-specific CaMKII inhibition with limited extracardiac effects.
In addition to CPVT, KN-93 has been shown to be effective in several other models of cardiac arrhythmias. Increased CaMKII-dependent SR Ca2+ leak is a hallmark of AF. Diastolic SR Ca2+ release via leaky RyR2 is hypothesized to contribute to arrhythmogenesis. In Ryr2R176Q/+ mice (exhibiting a gain-of-function defect in RyR), CaMKII inhibition prevented arrhythmias induced by rapid atrial pacing (Chelu et al., 2009). Studies have also indicated that KN-93 is effective at reversing the diastolic Ca2+ leak and attenuating the frequency of spontaneous Ca2+ transients in aged atrial murine myocytes (Guo et al., 2014), which is an important finding given that aging is associated with an increase risk of AF. Finally, CaMKII is also activated in HF and has been linked to HF-associated arrhythmias. In hypertrophic mice, KN-93 suppressed isoproterenol-induced ventricular arrhythmias and reduced isoproterenol-induced QTc prolongation (Feng et al., 2017). Furthermore, KN-93 significantly reduced arrhythmia inducibility and slowed initiation of ventricular tachycardia in a rabbit model of HF (Hoeker et al., 2016), strengthening the notion that CaMKII inhibition may have antiarrhythmic effects in the failing human heart.
W-7 has been identified as a CaMKII inhibitor; as a CaM antagonist it inhibits the Ca2+/CaM dependent activation of CaMKII thereby acting as an upstream blocker of CaMKII (Mazur et al., 1999). In a mouse model of cardiac hypertrophy (in which CaMKII expression levels were increased), inhibition of CaMKII activation by W-7 and KN-93 suppressed polymorphic ventricular arrhythmias (Figure 3). Here, W-7 was found to prolong AP duration to a greater extent than that of KN-93 (Kirchhof et al., 2004). W-7 has been shown to suppress TdP induction, without affecting QT duration or heart rate (Gbadebo et al., 2002; Pu et al., 2005). In a canine model of chronic AV block (associated with compensated hypertrophy and an increased risk of TdP) W-7 suppressed drug-induced TdP and shortened AP duration. Additional in vitro experiments found W-7 attenuated the dofetilide-induced enhanced CaMKII phosphorylation (Bourgonje et al., 2012).
TRP
Transient receptor potential channels may prove to be suitable therapeutic targets for arrhythmic disorders as a number of these channels have been implicated in arrhythmogenesis and they exhibit specific expression patterns in the cardiac conduction system (Hof et al., 2019). Within the SAN, TRP channels have been identified to contribute to both the membrane and Ca2+ clock (Hof et al., 2019). TRPC isoforms 1, 2, 3, 4, 6 and 7 have been detected in the mouse SAN, and treatment with the broad TRP channel inhibitor SKF-96365 results in reduced Ca2+ influx and a reduction in the spontaneous pacemaker rate (Ju et al., 2007). Store-operated Ca2+ entry (SOCE) describes the influx of Ca2+ into the cell when Ca2+ levels within the SR become depleted, a process which has been implicated in arrhythmia development (Bonilla et al., 2019). TRPC channels have been identified at store-operated transmembrane Ca2+ channels (Bonilla et al., 2019). With TRPC3 being the only TRPC channel to be located at the plasma membrane of pacemaker cells, it has been suggested that this channel is the main isoform to be involved in SOCE in the SAN (Hof et al., 2019). Spontaneous AP events via SOCE-activation were found to be reduced in a TRPC3 knockout mouse model (Ju et al., 2015), while excessive activation of TRPC3 has been shown to result in Ca2+ overload and arrhythmias. Under normal conditions, TRPC3 is understood to mediate Ca2+ and Na+ entry near NCX1, thereby promoting the forward mode of NCX and driving diastolic depolarization (Doleschal et al., 2015; Hof et al., 2019). However, when TRPC activation is increased, Ca2+ overload and spatial uncoupling between TRPC3 and NCX1 occurs, resulting in arrhythmogenesis (Doleschal et al., 2015). This evidence therefore suggests that TRPC3 contributes to SAN pacemaking through the stimulation of the Ca2+ clock via SOCE, activation of CaMKII and phosphorylation of Ca2+ handling proteins (including PLN and RyR2), and via TRPC3-NCX dynamics (Hof et al., 2019).
TRPC3 expression is upregulated in the atria of AF patients, and in numerous animal models of AF (Harada et al., 2012; Zhao et al., 2013), conversely, when TRPC3 expression is deleted, in a TRPC3 knockout mouse model, pacing-induced AF was significantly reduced (Ju et al., 2015). Further studies have suggested TRPC3 overexpression in AF mediates cardiac fibroblast proliferation and differentiation via the regulation of Ca2+ influx and downstream Ca2+-mediated signaling (Harada et al., 2012). In a canine model of AF, treatment with the selective TRPC3 blocker Pyr-3 reduced atrial fibroblast proliferation and therefore the development of an AF substrate (Figure 4) (Harada et al., 2012). TRPC3 has also been linked to other arrhythmia conditions in addition to AF. Its activation by GSK1702934A has been shown to influence cardiac contractility and favor rhythmic instability (Doleschal et al., 2015), and in a TRPC3 overexpression mouse model, GSK1702934A resulted in arrhythmic events including paired ventricular beats, and episodes of atrial tachycardia, ventricular tachycardia and cardiac alternans (Doleschal et al., 2015). TRPC3 overexpressing hearts were also more susceptible to ANG-II-stimulated arrhythmic events which included atrial or ventricular tachycardia. The interaction between TRPC3 and NCX1 is believed to drive the increased contractility and arrhythmogenesis upon TRPC3 activation (Doleschal et al., 2015). Therefore, inhibiting TRPC3 may be a potential therapeutic approach for arrhythmias arising due to sinus node dysfunction and AF. Pyr-3 has been identified as a selective inhibitor of TRPC3 and has been shown to efficiently suppress physiological events associated with TRPC3, including atrial fibroblast proliferation as highlighted above (Kiyonaka et al., 2009; Harada et al., 2012).
Studies have identified TRPM4 to be involved in SAN pacemaking through modulation of diastolic depolarization, suggesting that therapeutically targeting TRPM4 channels may be useful for heart rate modulation (Hof et al., 2019). Inhibition of TRPM4 by 9-phenanthrol reduced spontaneous AP in isolated mouse and rat atrial preparations and resulted in a frequency-dependent reduction of right atria beating rate (Figure 4 and Table 2). Similar results were evident in rabbit SAN pacemaker cells where inhibition of TRPM4 reduced the diastolic depolarization slope (Hof et al., 2013). The study suggested that TRPM4 may act as a rate accelerator channel in the SAN that becomes activated when heart rate decreases to provide protection against bradycardia (Hof et al., 2013). In addition to TRPM4, TRPM7 has also been detected in the mouse SAN (Hof et al., 2019) and is required for maintaining cardiac automaticity. Deletion of TRPM7 in vitro slows spontaneous Ca2+ transient frequency and disrupts cardiac automaticity, while in vivo a loss-of-function zebrafish model displayed a slowing of heart rate (Sah et al., 2013b). In addition, the study of two mouse models of TRPM7 deletion has revealed a role for TRPM7 SAN function: (1) a cardiomyocyte-specific deletion of TRPM7 resulted in sinus pauses and (2) an inducible SAN/AVN TRPM7 knock-out drove atrioventricular block. These phenotypes were associated with a reduction in diastolic depolarization due to a downregulation of HCN4, and If, as a result of TRPM7 deletion (Sah et al., 2013b).
Both TRPM4 and TRPM7 are also expressed in human atrial cells (Hof et al., 2019). Small animal studies have identified that these TRP channels contribute to the atrial AP (Simard et al., 2013), with inhibition of TRPM4 by 9-phenanthrol leading to a shortening of the atrial AP duration, suggesting it may be a potential target for pharmacological approaches against atrial arrhythmias (Simard et al., 2013). It is proposed that TRPM7 channels are involved in fibrogenesis in human AF, and TRPM7 protein expression is upregulated in atrial specimens from AF patients (Zhang et al., 2012). In terms of ventricle conduction, studies using a knockout TRPM4 mouse model found that its loss increased β-adrenergic inotropic response, with TRPM4–/– ventricular myocytes displaying a faster repolarization phase and a shorter AP waveform compared to controls (Mathar et al., 2014). The study concluded that TRPM4 reduces the driving force of Ca2+ influx, and subsequently contractile force, with the action of TRPM4 specifically important during β-adrenergic stimulation when there is an increase in the Ca2+ current. TRPM4 channels have also been identified in Purkinje fibers, with mRNA expression most abundant in the Purkinje fibers compared to the atria, ventricle and septum (Hof et al., 2019). Inhibition of TRPM4 by 9-phenanthrol was identified to shorten the AP of Purkinje fibers isolated from rabbit hearts with no effect on ventricle AP, suggesting TRPM4 is specifically involved in Purkinje fiber AP regulation (Hof et al., 2016).
The clinical importance of TRPM4 in cardiac conduction has been highlighted by studies that have identified TRPM4 mutations in numerous arrhythmic conditions including progressive cardiac bundle branch disease (Kruse et al., 2009), Brugada syndrome (Liu et al., 2013), LQT syndrome (Hof et al., 2017), atrioventricular and right bundle branch block (Stallmeyer et al., 2012), chronic heart block (Bianchi et al., 2018), and ventricular fibrillation (Bianchi et al., 2018). Furthermore, the cardiac phenotype of TRPM4 knockout mice included conduction block with PR ad QRS lengthening and Luciani-Wenckebach atrioventricular block (Demion et al., 2014). Interestingly, the mutations identified in familial arrhythmic conditions included loss- and gain- of function mutations, which suggests targets may have to be specific for the underlying mutation.
As highlighted in the studies above, 9-phenanthrol is a commonly used inhibitor of TRPM4 (Figure 4 and Table 2). The phenanthrene-derivative has been shown to be a specific inhibitor of TRPM4 channels in numerous tissues in the body (Guinamard et al., 2014). Within cardiomyocytes it is worth noting that 9-phenanthrol has been shown to modulate intracellular Ca2+ oscillation in HL-1 cells derived from mouse atrial cardiomyocytes, resulting in a transient increase in intracellular Ca2+ levels (most likely from mitochondrial Ca2+ stores). The study suggested this may be due to a feedback mechanism from inhibition of TRPM4 since it is known to be activated by Ca2+ (Burt et al., 2013). While 9-phenanthrol is a strong TRPM4 inhibitor for in vitro and ex vivo studies, the use of the agent in vivo is limited due low solubility and possible toxicity (Guinamard et al., 2014). In terms of TRPM7, pharmacological agents driving specific TRPM7 inhibition within the heart are lacking. TRPM7 is also one of several TRP channels which display broad tissue expression, suggesting targeting of this channel may result in off-target effects (Hof et al., 2019). Despite this, TRPM7 remains a potential therapeutic target for a range of conditions, including cardiac arrhythmias.
In addition to TRPC and TRPM channels, further TRP channels have been implicated in cardiac rhythm. Increased gene expression of various TRP channels (including TRPM, TRPC, TRPV, and TRPP) have been identified in leukocytes of patients with non-valvular AF (Duzen et al., 2017). TRPV channels have been linked to atrial inotropy related to ROS-mediated signaling (Odnoshivkina et al., 2015), and in a porcine model, depletion of TRPV1 afferent fibers resulted in a reduction in ventricular arrhythmic events associated with MI (Yoshie et al., 2020). A study has shown that TRPA1 may be a novel therapeutic target for increasing inotropic and lusitropic states in the heart, having demonstrated that stimulation of TRPA1 in isolated murine cardiomyocytes resulted in activation of CaMKII-dependent signaling and an increase in peak intracellular Ca2+ levels, driving an increase in cardiomyocyte contractile function (Andrei et al., 2017). Finally, in a zebrafish model, mutations in PKD2 (which encodes for TRPP2) resulted in atrioventricular block, with isolated hearts displaying impaired intracellular Ca2+ cycling and Ca2+ alternans (Paavola et al., 2013).
MCU
It has been suggested that cardiac mitochondria influence arrhythmia development through the regulation of cytoplasmic Ca2+ fluxes achieved via the uptake of Ca2+ through MCU (Brown and O’Rourke, 2010). Much of the work focused on targeting MCU as a potential therapeutic approach to arrhythmogenesis has focused on HF models. A study utilizing a knockout MCU mouse model found, in a non-ischemic HF setting, MCU knockout mice display a shorter AP duration and fewer incidences of EADs and ventricular fibrillation when compared to control animals (Xie et al., 2018). Use of the MCU blocker RR was found to reduce the incidence of ventricular fibrillation in an I/R rat model (Figure 4 and Table 2) (de et al., 2006). However, RR has off-target effects related to Ca2+ dynamics, including SR Ca2+ release and LTCC (Brown and O’Rourke, 2010). Ru360, an RR analog, has been shown to be a more specific MCU inhibitor and able to suppress ventricular fibrillation associated with I/R (Figure 4 and Table 2) (de et al., 2006). Both Ruthenium compounds have also been shown to convert pacing-induced ventricular fibrillation to ventricular tachycardia in isolated rat hearts, highlighting the role of MCU in arrhythmia development (Kawahara et al., 2003). It is worth noting that while Ru360 had no effect on left ventricle pressure, at high concentrations RR reduced left ventricle pressure amplitude (Kawahara et al., 2003).
Discussion
Ca2+ plays a critical role in maintaining cardiac physiology and changes to the proteins which are involved in Ca2+ handling underly numerous cardiac diseases. Targeting Ca2+ dysregulation in HF and arrhythmias has long been investigated as a potential therapeutic target and, as is evident from this review, there is considerable current research investigating targeting proteins associated with Ca2+ handling, including RyR2, NCX, SERCA, and TRP channels, as a therapeutic approach for cardiomyopathy, HF and arrhythmias. The pathogenic remodeling of these Ca2+ handling proteins has been shown to differ dependent on the underlying pathological mechanism, suggesting a “personalized” approach to targeting Ca2+ handling proteins may be required for specific cardiac conditions. Furthermore, while several therapeutics targeting Ca2+ have progressed to clinical trials, several are still in early preclinical stages, demonstrating the need for further research in this area. Altogether, targeting Ca2+ handling is a promising approach to developing novel and effective therapies for key cardiovascular disorders.
Author Contributions
EC, CW, and AN contributed conceptually to the manuscript. AN and CW drafted the manuscript. AN contributed the section concerning cardiac hypertrophy and heart failure, and the tables. CW contributed the section regarding cardiac arrhythmias. Both AN and CW contributed to the introduction, the current therapeutic approaches, the discussion, the figures, and the abstract. All authors reviewed the manuscript prior to submission.
Funding
This study was supported by the British Heart Foundation (FS/13/56/30645) and the Medical Research Council (G1002082).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
AC, adenylyl cyclase; AC6, adenylyl cyclase 6; ACE, angiotensin-converting enzyme; ACEI, ACE inhibitors; AF, atrial fibrillation; ANG-II, angiotensin II; AP, action potential; ARB, angiotensin receptor blocker; AVN, atrioventricular node; Ca2+, calcium; CaM, calmodulin; CaMKII, Ca2+/calmodulin-dependent protein kinase; CPVT, catecholaminergic polymorphic ventricular tachycardia; CVD, cardiovascular disease; DAD, delayed after depolarization; EAD, early after depolarization; ECC, excitation contraction coupling; GWAS, genome wide association studies; HF, heart failure; HFmEF, HF with mid-range ejection fraction; HFpEF, HF with preserved ejection fraction; HFrEF, HF with reduced ejection fraction; HNO/NO, nitroxyl; ICa, Ca2+ current; ICaL, L-type Ca2+ current; IK, K+ current; INa, Na+ current; INCX, NCX current; Ito, transient outward K+ current; I/R, ischemia/reperfusion; LQTS, long QT syndrome; LTCC, L-type Ca2+ channel; MAPK, mitogen activated protein kinase; MCU, mitochondrial Ca2+ uniporter; MI, myocardial infarction; NCX, Na+/Ca2+ exchanger; NFAT, nuclear factor of activated T-cell; PKA, protein kinase A; PLN, phospholamban; PMCA, plasma membrane Ca2+ ATPase; Pyr-3, pyrazole-3; RAAS, renin–angiotensin–aldosterone signaling; ROS, reactive oxygen species; RR, ruthenium red; RyR, ryanodine receptor; SAN, sino-atrial node; SERCA, SR Ca2+ ATPase; SNPs, single nucleotide polymorphisms; SR, sarcoplasmic reticulum; TAC, transverse aortic constriction; TdP, Torsades de Pointes; TRIC, trimeric intracellular cation.
References
Abu-Zeitone, A., Peterson, D. R., Polonsky, B., McNitt, S., and Moss, A. J. (2014). Efficacy of different beta-blockers in the treatment of long QT syndrome. J. Am. Coll. Cardiol. 64, 1352–1358. doi: 10.1016/j.jacc.2014.05.068
Adachi, T., Weisbrod, R. M., Pimentel, D. R., Ying, J., Sharov, V. S., Schoneich, C., et al. (2004). S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat. Med. 10, 1200–1207. doi: 10.1038/nm1119
Al-Khatib, S. M., Stevenson, W. G., Ackerman, M. J., Bryant, W. J., Callans, D. J., Curtis, A. B., et al. (2018). 2017 AHA/ACC/HRS Guideline for Management of Patients With Ventricular Arrhythmias and the prevention of sudden cardiac death: executive summary: a report of the American College of Cardiology/American Heart Association task force on clinical practice guidelines and the heart rhythm society. J. Am. Coll. Cardiol. 72, 1677–1749.
Al-Mohammad, A., Mant, J., Laramee, P., and Swain, S. Chronic Heart Failure Guideline Development G. (2010). Diagnosis and management of adults with chronic heart failure: summary of updated NICE guidance. BMJ 341:c4130. doi: 10.1136/bmj.c4130
Anderson, M. E. (2009). CaMKII and a failing strategy for growth in heart. J. Clin. Invest. 119, 1082–1085. doi: 10.1172/jci39262
Andersson, D. C., and Marks, A. R. (2010). Fixing ryanodine receptor Ca leak - a novel therapeutic strategy for contractile failure in heart and skeletal muscle. Drug Discov. Today Dis. Mech. 7, e151–e157. doi: 10.1016/j.ddmec.2010.09.009
Andersson, K. B., Birkeland, J. A., Finsen, A. V., Louch, W. E., Sjaastad, I., Wang, Y., et al. (2009). Moderate heart dysfunction in mice with inducible cardiomyocyte-specific excision of the Serca2 gene. J. Mol. Cell Cardiol. 47, 180–187. doi: 10.1016/j.yjmcc.2009.03.013
Andrei, S. R., Ghosh, M., Sinharoy, P., Dey, S., Bratz, I. N., and Damron, D. S. (2017). TRPA1 ion channel stimulation enhances cardiomyocyte contractile function via a CaMKII-dependent pathway. Channels 11, 587–603. doi: 10.1080/19336950.2017.1365206
Andrikopoulos, G. K., Pastromas, S., and Tzeis, S. (2015). Flecainide: current status and perspectives in arrhythmia management. World J. Cardiol. 7, 76–85.
Antoons, G., and Sipido, K. R. (2008). Targeting calcium handling in arrhythmias. Europace 10, 1364–1369. doi: 10.1093/europace/eun271
Antos, C. L., Frey, N., Marx, S. O., Reiken, S., Gaburjakova, M., Richardson, J. A., et al. (2001). Dilated cardiomyopathy and sudden death resulting from constitutive activation of protein kinase a. Circ. Res. 89, 997–1004. doi: 10.1161/hh2301.100003
Antzelevitch, C., and Burashnikov, A. (2011). Overview of Basic Mechanisms of Cardiac Arrhythmia. Card Electrophysiol. Clin. 3, 23–45. doi: 10.1016/j.ccep.2010.10.012
Armesilla, A. L., Williams, J. C., Buch, M. H., Pickard, A., Emerson, M., Cartwright, E. J., et al. (2004). Novel functional interaction between the plasma membrane Ca2+ pump 4b and the proapoptotic tumor suppressor Ras-associated factor 1 (RASSF1). J. Biol. Chem. 279, 31318–31328. doi: 10.1074/jbc.m307557200
Ashna, A., van Helden, D. F., Dos Remedios, C., Molenaar, P., and Laver, D. R. (2020). Phenytoin Reduces Activity of Cardiac Ryanodine Receptor 2; A Potential Mechanism for Its Cardioprotective Action. Mol. Pharmacol. 97, 250–258. doi: 10.1124/mol.119.117721
Backs, J., Backs, T., Neef, S., Kreusser, M. M., Lehmann, L. H., Patrick, D. M., et al. (2009). The delta isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload. Proc. Natl. Acad. Sci. U.S.A. 106, 2342–2347. doi: 10.1073/pnas.0813013106
Balam Ortiz, E. O., Carvajal, K., and Cruz, D. (1999). [Protective effect of dantrolene in post-ischemic reperfusion myocardial damage]. Arch. Inst. Cardiol. Mex. 69, 311–319.
Beauverger, P., Ozoux, M. L., Begis, G., Glenat, V., Briand, V., Philippo, M. C., et al. (2020). Reversion of cardiac dysfunction by a novel orally available calcium/calmodulin-dependent protein kinase II inhibitor. RA306, in a genetic model of dilated cardiomyopathy. Cardiovasc. Res. 116, 329–338.
Bers, D. M. (2014). Cardiac sarcoplasmic reticulum calcium leak: basis and roles in cardiac dysfunction. Annu. Rev. Physiol. 76, 107–127. doi: 10.1146/annurev-physiol-020911-153308
Bers, D. M., and Despa, S. (2006). Cardiac myocytes Ca2+ and Na+ regulation in normal and failing hearts. J. Pharmacol. Sci. 100, 315–322. doi: 10.1254/jphs.cpj06001x
Beuckelmann, D. J., Nabauer, M., and Erdmann, E. (1992). Intracellular calcium handling in isolated ventricular myocytes from patients with terminal heart failure. Circulation 85, 1046–1055. doi: 10.1161/01.cir.85.3.1046
Beutner, G., Sharma, V. K., Giovannucci, D. R., Yule, D. I., and Sheu, S. S. (2001). Identification of a ryanodine receptor in rat heart mitochondria. J. Biol. Chem. 276, 21482–21488. doi: 10.1074/jbc.m101486200
Bezzerides, V. J., Caballero, A., Wang, S., Ai, Y., Hylind, R. J., Lu, F., et al. (2019). Gene Therapy for Catecholaminergic Polymorphic Ventricular Tachycardia by Inhibition of Ca(2+)/Calmodulin-Dependent Kinase II. Circulation 140, 405–419. doi: 10.1161/circulationaha.118.038514
Bianchi, B., Ozhathil, L. C., Medeiros-Domingo, A., Gollob, M. H., and Abriel, H. (2018). Four TRPM4 cation channel mutations found in cardiac conduction diseases lead to altered protein stability. Front. Physiol. 9:177. doi: 10.3389/fphys.2018.00177
Billman, G. E., and Hamlin, R. L. (1996). The effects of mibefradil, a novel calcium channel antagonist on ventricular arrhythmias induced by myocardial ischemia and programmed electrical stimulation. J. Pharmacol. Exp. Ther. 277, 1517–1526.
Bogeholz, N., Schulte, J. S., Kaese, S., Bauer, B. K., Pauls, P., Dechering, D. G., et al. (2017). The Effects of SEA0400 on Ca(2+) Transient Amplitude and Proarrhythmia Depend on the Na(+)/Ca(2+) exchanger expression level in murine models. Front. Pharmacol. 8:649. doi: 10.3389/fphar.2017.00649
Bonilla, I. M., Belevych, A. E., Baine, S., Stepanov, A., Mezache, L., Bodnar, T., et al. (2019). Enhancement of Cardiac Store Operated Calcium Entry (SOCE) within Novel Intercalated Disk Microdomains in Arrhythmic Disease. Sci. Rep. 9:10179.
Bossu, A., Kostense, A., Beekman, H. D. M., Houtman, M. J. C., van der Heyden, M. A. G., and Vos, M. A. (2018). Istaroxime, a positive inotropic agent devoid of proarrhythmic properties in sensitive chronic atrioventricular block dogs. Pharmacol. Res. 133, 132–140. doi: 10.1016/j.phrs.2018.05.001
Bourgonje, V. J., Schoenmakers, M., Beekman, J. D., van der Nagel, R., Houtman, M. J., Miedema, L. F., et al. (2012). Relevance of calmodulin/CaMKII activation for arrhythmogenesis in the AV block dog. Heart Rhythm. 9, 1875–1883.
Boyett, M. R. (2009). ‘And the beat goes on.’ The cardiac conduction system: the wiring system of the heart. Exp. Physiol. 94, 1035–1049. doi: 10.1113/expphysiol.2009.046920
Boys, J. A., Toledo, A. H., Anaya-Prado, R., Lopez-Neblina, F., and Toledo-Pereyra, L. H. (2010). Effects of dantrolene on ischemia-reperfusion injury in animal models: a review of outcomes in heart, brain, liver, and kidney. J. Investig. Med. 58, 875–882. doi: 10.2310/jim.0b013e3181e5d719
Brenner, J. S., and Dolmetsch, R. E. (2007). TrpC3 regulates hypertrophy-associated gene expression without affecting myocyte beating or cell size. PLoS One 2:e802. doi: 10.1371/journal.pone.0000802
Brogden, R. N., and Markham, A. (1997). Mibefradil. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic efficacy in the management of hypertension and angina pectoris. Drugs 54, 774–793. doi: 10.2165/00003495-199754050-00010
Brown, D. A., and O’Rourke, B. (2010). Cardiac mitochondria and arrhythmias. Cardiovasc. Res. 88, 241–249. doi: 10.1093/cvr/cvq231
Bueno, O. F., De Windt, L. J., Tymitz, K. M., Witt, S. A., Kimball, T. R., Klevitsky, R., et al. (2000). The MEK1-ERK1/2 signaling pathway promotes compensated cardiac hypertrophy in transgenic mice. EMBO J. 19, 6341–6350. doi: 10.1093/emboj/19.23.6341
Bursztyn, M., Kadr, H., Tilvis, R., Martina, B., Oigman, W., Talberg, J., et al. (1997). Mibefradil, a novel calcium antagonist, in elderly patients with hypertension: favorable hemodynamics and pharmacokinetics. Am. Heart J. 134, 238–247. doi: 10.1016/s0002-8703(97)70130-3
Burt, R., Graves, B. M., Gao, M., Li, C., Williams, D. L., Fregoso, S. P., et al. (2013). 9-Phenanthrol and flufenamic acid inhibit calcium oscillations in HL-1 mouse cardiomyocytes. Cell Calcium 54, 193–201. doi: 10.1016/j.ceca.2013.06.003
Bush, E. W., Hood, D. B., Papst, P. J., Chapo, J. A., Minobe, W., Bristow, M. R., et al. (2006). Canonical transient receptor potential channels promote cardiomyocyte hypertrophy through activation of calcineurin signaling. J. Biol. Chem. 281, 33487–33496. doi: 10.1074/jbc.m605536200
Campbell, T. J., and MacDonald, P. S. (2003). Digoxin in heart failure and cardiac arrhythmias. Med. J. Aust. 179, 98–102. doi: 10.5694/j.1326-5377.2003.tb05445.x
Cardiac Arrhythmia Suppression Trial I. (1989). Preliminary report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. N. Engl. J. Med. 321, 406–412. doi: 10.1056/nejm198908103210629
Cartwright, E. J., Oceandy, D., Austin, C., and Neyses, L. (2011). Ca2+ signalling in cardiovascular disease: the role of the plasma membrane calcium pumps. Sci. China-Life Sci. 54, 691–698. doi: 10.1007/s11427-011-4199-1
Chang, P. C., Wo, H. T., Lee, H. L., Wen, M. S., and Chou, C. C. (2015). Paradoxical effects of KB-R7943 on arrhythmogenicity in a chronic myocardial infarction rabbit model. J. Cardiol. 66, 80–87. doi: 10.1016/j.jjcc.2014.08.002
Chatterjee, S., Biondi-Zoccai, G., Abbate, A., D’Ascenzo, F., Castagno, D., Van Tassell, B., et al. (2013). Benefits of beta blockers in patients with heart failure and reduced ejection fraction: network meta-analysis. BMJ 346:f55.
Chelu, M. G., Sarma, S., Sood, S., Wang, S., van Oort, R. J., Skapura, D. G., et al. (2009). Calmodulin kinase II-mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. J. Clin. Invest. 119, 1940–1951.
Chevalier, M., Vermij, S. H., Wyler, K., Gillet, L., Keller, I., and Abriel, H. (2018). Transcriptomic analyses of murine ventricular cardiomyocytes. Sci. Data 5:180170.
Chockalingam, P., Crotti, L., Girardengo, G., Johnson, J. N., Harris, K. M., van der Heijden, J. F., et al. (2012). Not all beta-blockers are equal in the management of long QT syndrome types 1 and 2: higher recurrence of events under metoprolol. J. Am. Coll. Cardiol. 60, 2092–2099.
CIBIS-II Investigators and Committees (1999). The Cardiac Insufficiency Bisoprolol Study II (CIBIS-II): a randomised trial. Lancet 353, 9–13 doi: 10.1016/s0140-6736(98)11181-9
Collaborators GBDCoD. (2018). Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 392, 1736–1788.
Correll, R. N., Makarewich, C. A., Zhang, H., Zhang, C., Sargent, M. A., York, A. J., et al. (2017). Caveolae-localized L-type Ca2+ channels do not contribute to function or hypertrophic signalling in the mouse heart. Cardiovasc. Res. 113, 749–759. doi: 10.1093/cvr/cvx046
Cowart, D., Venuti, R. P., Lynch, K., Guptill, J. T., Noveck, R. J., and Foo, S. Y. (2019). A Phase 1 Randomized Study of Single Intravenous Infusions of the Novel Nitroxyl Donor BMS-986231 in Healthy Volunteers. J. Clin. Pharmacol. 59, 717–730. doi: 10.1002/jcph.1364
Cutler, M. J., Wan, X., Plummer, B. N., Liu, H., Deschenes, I., Laurita, K. R., et al. (2012). Targeted sarcoplasmic reticulum Ca2+ ATPase 2a gene delivery to restore electrical stability in the failing heart. Circulation 126, 2095–2104. doi: 10.1161/circulationaha.111.071480
Dan, G. A., Martinez-Rubio, A., Agewall, S., Boriani, G., Borggrefe, M., Gaita, F., et al. (2018). Antiarrhythmic drugs-clinical use and clinical decision making: a consensus document from the European Heart Rhythm Association (EHRA) and European Society of Cardiology (ESC) Working Group on Cardiovascular Pharmacology, endorsed by the Heart Rhythm Society (HRS), Asia-Pacific Heart Rhythm Society (APHRS) and International Society of Cardiovascular Pharmacotherapy (ISCP). Europace 20, 731an–732an.
de Godoy, C. M. G., Vasques, E. R., Caricati-Neto, A., Tavares, J. G. P., Alves, B. J., Duarte, J., et al. (2018). Heparin oligosaccharides have antiarrhythmic effect by accelerating the sodium-calcium exchanger. Front. Cardiovasc. Med. 5:67. doi: 10.3389/fcvm.2018.00067
de, J., Garcia-Rivas, G., Carvajal, K., Correa, F., and Zazueta, C. (2006). Ru360, a specific mitochondrial calcium uptake inhibitor, improves cardiac post-ischaemic functional recovery in rats in vivo. Br. J. Pharmacol. 149, 829–837. doi: 10.1038/sj.bjp.0706932
del Monte, F., Harding, S. E., Schmidt, U., Matsui, T., Kang, Z. B., Dec, G. W., et al. (1999). Restoration of contractile function in isolated cardiomyocytes from failing human hearts by gene transfer of SERCA2a. Circulation 100, 2308–2311. doi: 10.1161/01.cir.100.23.2308
del Monte, F., Lebeche, D., Guerrero, J. L., Tsuji, T., Doye, A. A., Gwathmey, J. K., et al. (2004). Abrogation of ventricular arrhythmias in a model of ischemia and reperfusion by targeting myocardial calcium cycling. Proc. Natl. Acad. Sci. U.S.A. 101, 5622–5627. doi: 10.1073/pnas.0305778101
del Monte, F., Williams, E., Lebeche, D., Schmidt, U., Rosenzweig, A., Gwathmey, J. K., et al. (2001). Improvement in survival and cardiac metabolism after gene transfer of sarcoplasmic reticulum Ca(2+)-ATPase in a rat model of heart failure. Circulation 104, 1424–1429. doi: 10.1161/hc3601.095574
Demers, C., Mody, A., Teo, K. K., and McKelvie, R. S. (2005). ACE inhibitors in heart failure: what more do we need to know? Am. J. Cardiovasc. Drugs 5, 351–359. doi: 10.2165/00129784-200505060-00002
Demion, M., Thireau, J., Gueffier, M., Finan, A., Khoueiry, Z., Cassan, C., et al. (2014). Trpm4 gene invalidation leads to cardiac hypertrophy and electrophysiological alterations. PLoS One 9:e115256. doi: 10.1371/journal.pone.0115256
Deo, M., Weinberg, S. H., and Boyle, P. M. (2017). Calcium Dynamics and Cardiac Arrhythmia. Clin. Med. Insights Cardiol. 11:1179546817739523.
Di Pasquale, E., Lodola, F., Miragoli, M., Denegri, M., Avelino-Cruz, J. E., Buonocore, M., et al. (2013). CaMKII inhibition rectifies arrhythmic phenotype in a patient-specific model of catecholaminergic polymorphic ventricular tachycardia. Cell Death Dis. 4:e843. doi: 10.1038/cddis.2013.369
Doleschal, B., Primessnig, U., Wolkart, G., Wolf, S., Schernthaner, M., Lichtenegger, M., et al. (2015). TRPC3 contributes to regulation of cardiac contractility and arrhythmogenesis by dynamic interaction with NCX1. Cardiovasc. Res. 106, 163–173. doi: 10.1093/cvr/cvv022
Dominguez-Rodriguez, A., Mayoral-Gonzalez, I., Avila-Medina, J., de Rojas-de Pedro, E. S., Calderon-Sanchez, E., Diaz, I., et al. (2018). Urocortin-2 Prevents Dysregulation of Ca(2+) homeostasis and improves early cardiac remodeling after ischemia and reperfusion. Front. Physiol. 9:813. doi: 10.3389/fphys.2018.00813
Dong, Q., Li, J., Wu, Q. F., Zhao, N., Qian, C., Ding, D., et al. (2017). Blockage of transient receptor potential vanilloid 4 alleviates myocardial ischemia/reperfusion injury in mice. Sci. Rep. 7:42678.
Dragun, M., Gazova, A., Kyselovic, J., Hulman, M., and Matus, M. (2019). TRP channels expression profile in human end-stage heart failure. Medicina 55:380. doi: 10.3390/medicina55070380
Du, X. J., Cole, T. J., Tenis, N., Gao, X. M., Kontgen, F., Kemp, B. E., et al. (2002). Impaired cardiac contractility response to hemodynamic stress in S100A1-deficient mice. Mol. Cell Biol. 22, 2821–2829. doi: 10.1128/mcb.22.8.2821-2829.2002
Durham, J. A., Gandolfi, A. J., and Bentley, J. B. (1984). Hepatotoxicological evaluation of dantrolene sodium. Drug Chem. Toxicol. 7, 23–40. doi: 10.3109/01480548409014171
Duzen, I. V., Yavuz, F., Vuruskan, E., Saracoglu, E., Poyraz, F., Goksuluk, H., et al. (2017). Leukocyte TRP channel gene expressions in patients with non-valvular atrial fibrillation. Sci. Rep. 7:9272.
Dzau, V. J., Colucci, W. S., Hollenberg, N. K., and Williams, G. H. (1981). Relation of the renin-angiotensin-aldosterone system to clinical state in congestive heart failure. Circulation 63, 645–651. doi: 10.1161/01.cir.63.3.645
Elias, C. L., Lukas, A., Shurraw, S., Scott, J., Omelchenko, A., Gross, G. J., et al. (2001). Inhibition of Na+/Ca2+ exchange by KB-R7943: transport mode selectivity and antiarrhythmic consequences. Am. J. Physiol. Heart Circ. Physiol. 281, H1334–H1345.
Entin-Meer, M., Cohen, L., Hertzberg-Bigelman, E., Levy, R., Ben-Shoshan, J., and Keren, G. (2017). TRPV2 knockout mice demonstrate an improved cardiac performance following myocardial infarction due to attenuated activity of peri-infarct macrophages. PLoS One 12:e0177132. doi: 10.1371/journal.pone.0177132
Falcon, D., Galeano-Otero, I., Calderon-Sanchez, E., Del Toro, R., Martin-Bornez, M., Rosado, J. A., et al. (2019). TRP channels: current perspectives in the adverse cardiac remodeling. Front. Physiol. 10:159. doi: 10.3389/fphys.2019.00159
Farah, C., Michel, L. Y. M., and Balligand, J. L. (2018). Nitric oxide signalling in cardiovascular health and disease. Nat. Rev. Cardiol. 15, 292–316. doi: 10.1038/nrcardio.2017.224
Felker, G. M., Borentain, M., Cleland, J. G., DeSouza, M. M., Kessler, P. D., O’Connor, C. M., et al. (2019). Rationale and design for the development of a novel nitroxyl donor in patients with acute heart failure. Eur. J. Heart Fail. 21, 1022–1031. doi: 10.1002/ejhf.1504
Feng, Y., Cheng, J., Wei, B., and Wang, Y. (2017). CaMKII inhibition reduces isoproterenol-induced ischemia and arrhythmias in hypertrophic mice. Oncotarget 8, 17504–17509. doi: 10.18632/oncotarget.15099
Fernandez-Sanz, C., Ruiz-Meana, M., Miro-Casas, E., Nunez, E., Castellano, J., Loureiro, M., et al. (2014). Defective sarcoplasmic reticulum-mitochondria calcium exchange in aged mouse myocardium. Cell Death Dis. 5:e1573. doi: 10.1038/cddis.2014.526
Ferrandi, M., Barassi, P., Tadini-Buoninsegni, F., Bartolommei, G., Molinari, I., Tripodi, M. G., et al. (2013). Istaroxime stimulates SERCA2a and accelerates calcium cycling in heart failure by relieving phospholamban inhibition. Br. J. Pharmacol. 169, 1849–1861. doi: 10.1111/bph.12278
Fink, M., Noble, P. J., and Noble, D. (2011). Ca(2)(+)-induced delayed afterdepolarizations are triggered by dyadic subspace Ca2(2)(+) affirming that increasing SERCA reduces aftercontractions. Am. J. Physiol. Heart Circ. Physiol. 301, H921–H935.
Freichel, M., Berlin, M., Schurger, A., Mathar, I., Bacmeister, L., Medert, R., et al. (2017). “TRP Channels in the Heart,” in Neurobiology of TRP Channels, ed. T. L. R. Emir (Boca Raton, FL: CRC Press), 149–185.
Froehlich, J. P., Mahaney, J. E., Keceli, G., Pavlos, C. M., Goldstein, R., Redwood, A. J., et al. (2008). Phospholamban thiols play a central role in activation of the cardiac muscle sarcoplasmic reticulum calcium pump by nitroxyl. Biochemistry 47, 13150–13152. doi: 10.1021/bi801925p
Fukuta, H., Goto, T., Wakami, K., and Ohte, N. (2017). The effect of beta-blockers on mortality in heart failure with preserved ejection fraction: a meta-analysis of observational cohort and randomized controlled studies. Int. J. Cardiol. 228, 4–10. doi: 10.1016/j.ijcard.2016.11.239
Fukuto, J. M. (2019). A recent history of nitroxyl chemistry, pharmacology and therapeutic potential. Br. J. Pharmacol. 176, 135–146. doi: 10.1111/bph.14384
Galindo, C. L., Ryzhov, S., and Sawyer, D. B. (2014). Neuregulin as a heart failure therapy and mediator of reverse remodeling. Curr. Heart Fail Rep. 11, 40–49. doi: 10.1007/s11897-013-0176-2
Gandhi, A., Siedlecka, U., Shah, A. P., Navaratnarajah, M., Yacoub, M. H., and Terracciano, C. M. (2013). The effect of SN-6, a novel sodium-calcium exchange inhibitor, on contractility and calcium handling in isolated failing rat ventricular myocytes. Cardiovasc. Ther. 31, e115–e124. doi: 10.1111/1755-5922.12045
Gao, M. H., Tang, T., Guo, T., Sun, S. Q., Feramisco, J. R., and Hammond, H. K. (2004). Adenylyl cyclase type VI gene transfer reduces phospholamban expression in cardiac myocytes via activating transcription factor 3. J. Biol. Chem. 279, 38797–38802. doi: 10.1074/jbc.m405701200
Gao, R., Zhang, J., Cheng, L., Wu, X., Dong, W., Yang, X., et al. (2010). A Phase II, randomized, double-blind, multicenter, based on standard therapy, placebo-controlled study of the efficacy and safety of recombinant human neuregulin-1 in patients with chronic heart failure. J. Am. Coll. Cardiol. 55, 1907–1914. doi: 10.1016/j.jacc.2009.12.044
Gardner, R. T., Ripplinger, C. M., Myles, R. C., and Habecker, B. A. (2016). Molecular mechanisms of sympathetic remodeling and arrhythmias. Circ. Arrhythm. Electrophysiol. 9:e001359.
Gbadebo, T. D., Trimble, R. W., Khoo, M. S., Temple, J., Roden, D. M., and Anderson, M. E. (2002). Calmodulin inhibitor W-7 unmasks a novel electrocardiographic parameter that predicts initiation of torsade de pointes. Circulation 105, 770–774. doi: 10.1161/hc0602.103724
Ghali, J. K., Smith, W. B., Torre-Amione, G., Haynos, W., Rayburn, B. K., Amato, A., et al. (2007). A phase 1-2 dose-escalating study evaluating the safety and tolerability of istaroxime and specific effects on electrocardiographic and hemodynamic parameters in patients with chronic heart failure with reduced systolic function. Am. J. Cardiol. 99, 47A–56A.
Glaser, S., Steinbach, M., Opitz, C., Wruck, U., and Kleber, F. X. (2001). Torsades de pointes caused by Mibefradil. Eur. J. Heart Fail. 3, 627–630. doi: 10.1016/s1388-9842(01)00159-3
Godfraind, T. (2017). Discovery and development of calcium channel blockers. Front. Pharmacol. 8:286. doi: 10.3389/fphar.2017.00286
Goldhaber, J. I., and Hamilton, M. A. (2010). Role of inotropic agents in the treatment of heart failure. Circulation 121, 1655–1660. doi: 10.1161/circulationaha.109.899294
Goldstein, R. E., Boccuzzi, S. J., Cruess, D., and Nattel, S. (1991). Diltiazem increases late-onset congestive heart failure in postinfarction patients with early reduction in ejection fraction. The Adverse Experience Committee; and the Multicenter Diltiazem Postinfarction Research Group. Circulation 83, 52–60. doi: 10.1161/01.cir.83.1.52
Goonasekera, S. A., Hammer, K., Auger-Messier, M., Bodi, I., Chen, X., Zhang, H., et al. (2012). Decreased cardiac L-type Ca(2)(+) channel activity induces hypertrophy and heart failure in mice. J. Clin. Invest. 122, 280–290. doi: 10.1172/jci58227
Gorski, P. A., Jang, S. P., Jeong, D., Lee, A., Lee, P., Oh, J. G., et al. (2019). Role of SIRT1 in Modulating Acetylation of the Sarco-Endoplasmic Reticulum Ca(2+)-ATPase in Heart Failure. Circ. Res. 124, e63–e80.
Goyal, N., Skrdla, P., Schroyer, R., Kumar, S., Fernando, D., Oughton, A., et al. (2019). Clinical Pharmacokinetics, Safety, and Tolerability of a Novel, First-in-Class TRPV4 Ion Channel Inhibitor, GSK2798745, in Healthy and Heart Failure Subjects. Am. J. Cardiovasc. Drugs 19, 335–342. doi: 10.1007/s40256-018-00320-6
Greenberg, B., Butler, J., Felker, G. M., Ponikowski, P., Voors, A. A., Desai, A. S., et al. (2016). Calcium upregulation by percutaneous administration of gene therapy in patients with cardiac disease (CUPID 2): a randomised, multinational, double-blind, placebo-controlled, phase 2b trial. Lancet 387, 1178–1186. doi: 10.1016/s0140-6736(16)00082-9
Guinamard, R., Demion, M., Magaud, C., Potreau, D., and Bois, P. (2006). Functional expression of the TRPM4 cationic current in ventricular cardiomyocytes from spontaneously hypertensive rats. Hypertension 48, 587–594. doi: 10.1161/01.hyp.0000237864.65019.a5
Guinamard, R., Hof, T., and Del Negro, C. A. (2014). The TRPM4 channel inhibitor 9-phenanthrol. Br. J. Pharmacol. 171, 1600–1613. doi: 10.1111/bph.12582
Guo, T., Zhang, T., Mestril, R., and Bers, D. M. (2006). Ca2+/Calmodulin-dependent protein kinase II phosphorylation of ryanodine receptor does affect calcium sparks in mouse ventricular myocytes. Circ. Res. 99, 398–406. doi: 10.1161/01.res.0000236756.06252.13
Guo, X., Yuan, S., Liu, Z., and Fang, Q. (2014). Oxidation- and CaMKII-mediated sarcoplasmic reticulum Ca(2+) leak triggers atrial fibrillation in aging. J. Cardiovasc. Electrophysiol. 25, 645–652. doi: 10.1111/jce.12395
Gwathmey, J. K., Copelas, L., MacKinnon, R., Schoen, F. J., Feldman, M. D., Grossman, W., et al. (1987). Abnormal intracellular calcium handling in myocardium from patients with end-stage heart failure. Circ. Res. 61, 70–76. doi: 10.1161/01.res.61.1.70
Gwathmey, J. K., Warren, S. E., Briggs, G. M., Copelas, L., Feldman, M. D., Phillips, P. J., et al. (1991). Diastolic dysfunction in hypertrophic cardiomyopathy. Effect on active force generation during systole. J. Clin. Invest. 87, 1023–1031. doi: 10.1172/jci115061
Hammond, H. K., Penny, W. F., Traverse, J. H., Henry, T. D., Watkins, M. W., Yancy, C. W., et al. (2016). Intracoronary gene transfer of adenylyl cyclase 6 in patients with heart failure: a randomized clinical trial. JAMA Cardiol. 1, 163–171.
Harada, M., Luo, X., Qi, X. Y., Tadevosyan, A., Maguy, A., Ordog, B., et al. (2012). Transient receptor potential canonical-3 channel-dependent fibroblast regulation in atrial fibrillation. Circulation 126, 2051–2064. doi: 10.1161/circulationaha.112.121830
Hasenfuss, G., Pieske, B., Holubarsch, C., Alpert, N. R., and Just, H. (1993). Excitation-contraction coupling and contractile protein function in failing and nonfailing human myocardium. Adv. Exp. Med. Biol. 346, 91–100. doi: 10.1007/978-1-4615-2946-0_9
Hasenfuss, G., Schillinger, W., Lehnart, S. E., Preuss, M., Pieske, B., Maier, L. S., et al. (1999). Relationship between Na+-Ca2+-exchanger protein levels and diastolic function of failing human myocardium. Circulation 99, 641–648. doi: 10.1161/01.cir.99.5.641
Hayward, C., Banner, N. R., Morley-Smith, A., Lyon, A. R., and Harding, S. E. (2015). The current and future landscape of SERCA gene therapy for heart failure: a clinical perspective. Hum. Gene Ther. 26, 293–304. doi: 10.1089/hum.2015.018
Heart Outcomes Prevention Evaluation Study Investigator, Yusuf, S., Sleight, P., Pogue, J., Bosch, J., Davies, R., et al. (2000). Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. N. Engl. J. Med. 342, 145–153. doi: 10.1056/nejm200001203420301
Hobai, I. A., and O’Rourke, B. (2004). The potential of Na+/Ca2+ exchange blockers in the treatment of cardiac disease. Expert Opin. Invest. Drugs 13, 653–664. doi: 10.1517/13543784.13.6.653
Hoeker, G. S., Hanafy, M. A., Oster, R. A., Bers, D. M., and Pogwizd, S. M. (2016). Reduced Arrhythmia Inducibility With Calcium/Calmodulin-dependent Protein Kinase II Inhibition in Heart Failure Rabbits. J. Cardiovasc. Pharmacol. 67, 260–265. doi: 10.1097/fjc.0000000000000343
Hof, T., Chaigne, S., Recalde, A., Salle, L., Brette, F., and Guinamard, R. (2019). Transient receptor potential channels in cardiac health and disease. Nat. Rev. Cardiol. 16, 344–360. doi: 10.1038/s41569-018-0145-2
Hof, T., Liu, H., Salle, L., Schott, J. J., Ducreux, C., Millat, G., et al. (2017). TRPM4 non-selective cation channel variants in long QT syndrome. BMC Med. Genet. 18:31. doi: 10.1186/s12881-017-0397-4
Hof, T., Salle, L., Coulbault, L., Richer, R., Alexandre, J., Rouet, R., et al. (2016). TRPM4 non-selective cation channels influence action potentials in rabbit Purkinje fibres. J. Physiol. 594, 295–306. doi: 10.1113/jp271347
Hof, T., Simard, C., Rouet, R., Salle, L., and Guinamard, R. (2013). Implication of the TRPM4 nonselective cation channel in mammalian sinus rhythm. Heart Rhythm. 10, 1683–1689. doi: 10.1016/j.hrthm.2013.08.014
Huang, W., Rubinstein, J., Prieto, A. R., Thang, L. V., and Wang, D. H. (2009). Transient receptor potential vanilloid gene deletion exacerbates inflammation and atypical cardiac remodeling after myocardial infarction. Hypertension 53, 243–250. doi: 10.1161/hypertensionaha.108.118349
Huang, X., Song, Z., and Qu, Z. (2018). Determinants of early afterdepolarization properties in ventricular myocyte models. PLoS Comput. Biol. 14:e1006382. doi: 10.1371/journal.pcbi.1006382
Huke, S., and Knollmann, B. C. (2011). Oxidized CaMKII: a “heart stopper” for the sinus node? J. Clin. Invest. 121, 2975–2977. doi: 10.1172/jci58389
Hulot, J. S., Ishikawa, K., and Hajjar, R. J. (2016). Gene therapy for the treatment of heart failure: promise postponed. Eur. Heart J. 37, 1651–1658. doi: 10.1093/eurheartj/ehw019
Hulot, J. S., Salem, J. E., Redheuil, A., Collet, J. P., Varnous, S., Jourdain, P., et al. (2017). Effect of intracoronary administration of AAV1/SERCA2a on ventricular remodelling in patients with advanced systolic heart failure: results from the AGENT-HF randomized phase 2 trial. Eur. J. Heart Fail 19, 1534–1541. doi: 10.1002/ejhf.826
Imahashi, K., Pott, C., Goldhaber, J. I., Steenbergen, C., Philipson, K. D., and Murphy, E. (2005). Cardiac-specific ablation of the Na+-Ca2+ exchanger confers protection against ischemia/reperfusion injury. Circ. Res. 97, 916–921. doi: 10.1161/01.res.0000187456.06162.cb
Inserte, J., Garcia-Dorado, D., Ruiz-Meana, M., Padilla, F., Barrabes, J. A., Pina, P., et al. (2002). Effect of inhibition of Na(+)/Ca(2+) exchanger at the time of myocardial reperfusion on hypercontracture and cell death. Cardiovasc. Res. 55, 739–748. doi: 10.1016/s0008-6363(02)00461-3
Iravanian, S., and Dudley, S. C. Jr. (2008). The renin-angiotensin-aldosterone system (RAAS) and cardiac arrhythmias. Heart Rhythm. 5, S12–S17.
Irie, T., Sips, P. Y., Kai, S., Kida, K., Ikeda, K., Hirai, S., et al. (2015). S-Nitrosylation of calcium-handling proteins in cardiac adrenergic signaling and hypertrophy. Circ. Res. 117, 793–803. doi: 10.1161/circresaha.115.307157
Jabbour, A., Hayward, C. S., Keogh, A. M., Kotlyar, E., McCrohon, J. A., England, J. F., et al. (2011). Parenteral administration of recombinant human neuregulin-1 to patients with stable chronic heart failure produces favourable acute and chronic haemodynamic responses. Eur. J. Heart Fail. 13, 83–92. doi: 10.1093/eurjhf/hfq152
Jamshidi, J., Asnaashari, A., Alipoor, R., Mohammadi, S., Roostaei, S., Samadian, M. M., et al. (2018). ATP2B1 rs2681472 and STK39 rs35929607 polymorphisms and risk of Hypertension in Iranian Population. Med. J. Islam. Repub. Iran. 32:14.
January, C. T., Wann, L. S., Alpert, J. S., Calkins, H., Cigarroa, J. E., and Cleveland, J. C. Jr., et al. (2014). 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on practice guidelines and the Heart Rhythm Society. Circulation 130, e199–e267.
Jaruga-Killeen, E., Bull, D. A., Lotun, K., Henry, T. D., Egnaczyk, G., Reed, T. D., et al. (2019). Abstract 16296: Safety of First in Human Triple-Gene Therapy Candidate for Heart Failure Patients. Circulation 140, A16296–A16296.
Jaski, B. E., Jessup, M. L., Mancini, D. M., Cappola, T. P., Pauly, D. F., Greenberg, B., et al. (2009). Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID Trial), a first-in-human phase 1/2 clinical trial. J. Card. Fail 15, 171–181. doi: 10.1016/j.cardfail.2009.01.013
Jessup, M., Greenberg, B., Mancini, D., Cappola, T., Pauly, D. F., Jaski, B., et al. (2011). Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID): a phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+-ATPase in patients with advanced heart failure. Circulation 124, 304–313. doi: 10.1161/circulationaha.111.022889
Joung, B., Chen, P. S., and Lin, S. F. (2011). The role of the calcium and the voltage clocks in sinoatrial node dysfunction. Yonsei Med. J. 52, 211–219.
Ju, Y. K., Chu, Y., Chaulet, H., Lai, D., Gervasio, O. L., Graham, R. M., et al. (2007). Store-operated Ca2+ influx and expression of TRPC genes in mouse sinoatrial node. Circ. Res. 100, 1605–1614. doi: 10.1161/circresaha.107.152181
Ju, Y. K., Lee, B. H., Trajanovska, S., Hao, G., Allen, D. G., Lei, M., et al. (2015). The involvement of TRPC3 channels in sinoatrial arrhythmias. Front. Physiol. 6:86. doi: 10.3389/fphys.2015.00086
Kaneko, N., Matsuda, R., Hata, Y., and Shimamoto, K. (2009). Pharmacological characteristics and clinical applications of K201. Curr. Clin. Pharmacol. 4, 126–131. doi: 10.2174/157488409788184972
Kannankeril, P. J., Moore, J. P., Cerrone, M., Priori, S. G., Kertesz, N. J., Ro, P. S., et al. (2017). Efficacy of flecainide in the treatment of catecholaminergic polymorphic ventricular tachycardia: a randomized clinical trial. JAMA Cardiol. 2, 759–766.
Kannel, W. B., Dawber, T. R., Kagan, A., Revotskie, N., and Stokes, J. III (1961). Factors of risk in the development of coronary heart disease–six year follow-up experience. The Framingham Study. Ann. Intern. Med. 55, 33–50.
Kass, R. S., and Clancy, C. E. (2006). Basis and Treatment of Cardiac Arrhythmias. Berlin: Springer.
Kawahara, K., Takase, M., and Yamauchi, Y. (2003). Ruthenium red-induced transition from ventricular fibrillation to tachycardia in isolated rat hearts: possible involvement of changes in mitochondrial calcium uptake. Cardiovasc. Pathol. 12, 311–321. doi: 10.1016/s1054-8807(03)00090-5
Kawase, Y., Ly, H. Q., Prunier, F., Lebeche, D., Shi, Y., Jin, H., et al. (2008). Reversal of cardiac dysfunction after long-term expression of SERCA2a by gene transfer in a pre-clinical model of heart failure. J. Am. Coll. Cardiol. 51, 1112–1119. doi: 10.1016/j.jacc.2007.12.014
Keating, M. T., and Sanguinetti, M. C. (2001). Molecular and cellular mechanisms of cardiac arrhythmias. Cell 104, 569–580. doi: 10.1016/s0092-8674(01)00243-4
Kecskes, M., Jacobs, G., Kerselaers, S., Syam, N., Menigoz, A., Vangheluwe, P., et al. (2015). The Ca(2+)-activated cation channel TRPM4 is a negative regulator of angiotensin II-induced cardiac hypertrophy. Basic Res. Cardiol. 110:43.
Kehat, I., Davis, J., Tiburcy, M., Accornero, F., Saba-El-Leil, M. K., Maillet, M., et al. (2011). Extracellular signal-regulated kinases 1 and 2 regulate the balance between eccentric and concentric cardiac growth. Circ. Res. 108, 176–183. doi: 10.1161/circresaha.110.231514
Kho, C., Lee, A., Jeong, D., Oh, J. G., Chaanine, A. H., Kizana, E., et al. (2011). SUMO1-dependent modulation of SERCA2a in heart failure. Nature 477, 601–605. doi: 10.1038/nature10407
Kho, C., Lee, A., Jeong, D., Oh, J. G., Gorski, P. A., Fish, K., et al. (2015). Small-molecule activation of SERCA2a SUMOylation for the treatment of heart failure. Nat. Commun. 6:7229.
Khot, U. N., Khot, M. B., Bajzer, C. T., Sapp, S. K., Ohman, E. M., Brener, S. J., et al. (2003). Prevalence of conventional risk factors in patients with coronary heart disease. JAMA 290, 898–904.
Kieserman, J. M., Myers, V. D., Dubey, P., Cheung, J. Y., and Feldman, A. M. (2019). Current landscape of heart failure gene therapy. J. Am. Heart Assoc. 8:e012239.
Kirchhof, P., Fabritz, L., Kilic, A., Begrow, F., Breithardt, G., and Kuhn, M. (2004). Ventricular arrhythmias, increased cardiac calmodulin kinase II expression, and altered repolarization kinetics in ANP receptor deficient mice. J. Mol. Cell Cardiol. 36, 691–700. doi: 10.1016/j.yjmcc.2004.03.007
Kistamas, K., Veress, R., Horvath, B., Banyasz, T., Nanasi, P. P., and Eisner, D. A. (2020). Calcium handling defects and cardiac arrhythmia syndromes. Front Pharmacol. 11:72. doi: 10.3389/fphar.2020.00072
Kiyonaka, S., Kato, K., Nishida, M., Mio, K., Numaga, T., Sawaguchi, Y., et al. (2009). Selective and direct inhibition of TRPC3 channels underlies biological activities of a pyrazole compound. Proc. Natl. Acad. Sci. U.S.A. 106, 5400–5405. doi: 10.1073/pnas.0808793106
Kjekshus, J., Swedberg, K., and Snapinn, S. (1992). Effects of enalapril on long-term mortality in severe congestive heart failure. CONSENSUS Trial Group. Am. J. Cardiol. 69, 103–107. doi: 10.1016/0002-9149(92)90683-p
Kobayashi, S., Bannister, M. L., Gangopadhyay, J. P., Hamada, T., Parness, J., and Ikemoto, N. (2005). Dantrolene stabilizes domain interactions within the ryanodine receptor. J. Biol. Chem. 280, 6580–6587. doi: 10.1074/jbc.m408375200
Kobayashi, S., Wakeyama, T., Ono, S., Ikeda, Y., Omura, M., Oda, T., et al. (2020). A multicenter, randomized, double-blind, controlled study to evaluate the efficacy and safety of dantrolene on ventricular arrhythmia as well as mortality and morbidity in patients with chronic heart failure (SHO-IN trial): rationale and design. J. Cardiol. 75, 454–461. doi: 10.1016/j.jjcc.2019.08.020
Kobayashi, S., Yano, M., Suetomi, T., Ono, M., Tateishi, H., Mochizuki, M., et al. (2009). Dantrolene, a therapeutic agent for malignant hyperthermia, markedly improves the function of failing cardiomyocytes by stabilizing interdomain interactions within the ryanodine receptor. J. Am. Coll. Cardiol. 53, 1993–2005. doi: 10.1016/j.jacc.2009.01.065
Kobayashi, S., Yano, M., Uchinoumi, H., Suetomi, T., Susa, T., Ono, M., et al. (2010). Dantrolene, a therapeutic agent for malignant hyperthermia, inhibits catecholaminergic polymorphic ventricular tachycardia in a RyR2(R2474S/+) knock-in mouse model. Circ. J. 74, 2579–2584. doi: 10.1253/circj.cj-10-0680
Kobayashi, Y., Hirawa, N., Tabara, Y., Muraoka, H., Fujita, M., Miyazaki, N., et al. (2012). Mice Lacking Hypertension Candidate Gene ATP2B1 in Vascular Smooth Muscle Cells Show Significant Blood Pressure Elevation. Hypertension 59, 854–U213.
Kobrin, I., Charlon, V., Lindberg, E., and Pordy, R. (1997). Safety of mibefradil, a new once-a-day, selective T-type calcium channel antagonist. Am. J. Cardiol. 80, 40C–46C. doi: 10.1016/s0002-9149(97)00569-9
Kohajda, Z., Farkas-Morvay, N., Jost, N., Nagy, N., Geramipour, A., Horvath, A., et al. (2016). The effect of a novel highly selective inhibitor of the sodium/calcium exchanger (NCX) on cardiac arrhythmias in in vitro and in vivo experiments. PLoS One 11:e0166041. doi: 10.1371/journal.pone.0166041
Komuro, I., Kurabayashi, M., Shibazaki, Y., Takaku, F., and Yazaki, Y. (1989). Molecular cloning and characterization of a Ca2+ + Mg2+-dependent adenosine triphosphatase from rat cardiac sarcoplasmic reticulum. Regulation of its expression by pressure overload and developmental stage. J. Clin. Invest. 83, 1102–1108. doi: 10.1172/jci113989
Kranias, E. G., and Hajjar, R. J. (2012). Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ. Res. 110, 1646–1660. doi: 10.1161/circresaha.111.259754
Kraus, C., Rohde, D., Weidenhammer, C., Qiu, G., Pleger, S. T., Voelkers, M., et al. (2009). S100A1 in cardiovascular health and disease: closing the gap between basic science and clinical therapy. J. Mol. Cell Cardiol. 47, 445–455. doi: 10.1016/j.yjmcc.2009.06.003
Kruse, M., Schulze-Bahr, E., Corfield, V., Beckmann, A., Stallmeyer, B., Kurtbay, G., et al. (2009). Impaired endocytosis of the ion channel TRPM4 is associated with human progressive familial heart block type I. J. Clin. Invest. 119, 2737–2744. doi: 10.1172/jci38292
Kumagai, K., Nakashima, H., Gondo, N., and Saku, K. (2003). Antiarrhythmic effects of JTV-519, a novel cardioprotective drug, on atrial fibrillation/flutter in a canine sterile pericarditis model. J. Cardiovasc. Electrophysiol. 14, 880–884. doi: 10.1046/j.1540-8167.2003.03050.x
Kuwahara, K., Wang, Y., McAnally, J., Richardson, J. A., Bassel-Duby, R., Hill, J. A., et al. (2006). TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. J. Clin. Invest. 116, 3114–3126. doi: 10.1172/jci27702
Kwong, J. Q. (2017). The mitochondrial calcium uniporter in the heart: energetics and beyond. J. Physiol. 595, 3743–3751. doi: 10.1113/jp273059
Ladilov, Y., Haffner, S., Balser-Schafer, C., Maxeiner, H., and Piper, H. M. (1999). Cardioprotective effects of KB-R7943: a novel inhibitor of the reverse mode of Na+/Ca2+ exchanger. Am. J. Physiol. 276, H1868–H1876.
Lai, N. C., Roth, D. M., Gao, M. H., Tang, T., Dalton, N., Lai, Y. Y., et al. (2004). Intracoronary adenovirus encoding adenylyl cyclase VI increases left ventricular function in heart failure. Circulation 110, 330–336. doi: 10.1161/01.cir.0000136033.21777.4d
Landstrom, A. P., Dobrev, D., and Wehrens, X. H. T. (2017). Calcium Signaling and Cardiac Arrhythmias. Circ. Res. 120, 1969–1993. doi: 10.1161/circresaha.117.310083
Lazzeroni, D., Rimoldi, O., and Camici, P. G. (2016). From Left Ventricular Hypertrophy to Dysfunction and Failure. Circ. J. 80, 555–564. doi: 10.1253/circj.cj-16-0062
Lee, C., Visen, N. S., Dhalla, N. S., Le, H. D., Isaac, M., Choptiany, P., et al. (2004). Inhibitory profile of SEA0400 [2-[4-[(2,5-difluorophenyl)methoxy]phenoxy]-5-ethoxyaniline] assessed on the cardiac Na+-Ca2+ exchanger. NCX1.1. J. Pharmacol. Exp. Ther. 311, 748–757. doi: 10.1124/jpet.104.070805
Lehnart, S. E., Mongillo, M., Bellinger, A., Lindegger, N., Chen, B. X., Hsueh, W., et al. (2008). Leaky Ca2+ release channel/ryanodine receptor 2 causes seizures and sudden cardiac death in mice. J. Clin. Invest. 118, 2230–2245.
Lei, M., Wu, L., Terrar, D. A., and Huang, C. L. (2018). Modernized Classification of Cardiac Antiarrhythmic Drugs. Circulation 138, 1879–1896. doi: 10.1161/circulationaha.118.035455
Lemmens, K., Fransen, P., Sys, S. U., Brutsaert, D. L., and De Keulenaer, G. W. (2004). Neuregulin-1 induces a negative inotropic effect in cardiac muscle: role of nitric oxide synthase. Circulation 109, 324–326. doi: 10.1161/01.cir.0000114521.88547.5e
Leren, I. S., Saberniak, J., Majid, E., Haland, T. F., Edvardsen, T., and Haugaa, K. H. (2016). Nadolol decreases the incidence and severity of ventricular arrhythmias during exercise stress testing compared with beta1-selective beta-blockers in patients with catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 13, 433–440. doi: 10.1016/j.hrthm.2015.09.029
Li, L., Louch, W. E., Niederer, S. A., Aronsen, J. M., Christensen, G., Sejersted, O. M., et al. (2012). Sodium accumulation in SERCA knockout-induced heart failure. Biophys. J. 102, 2039–2048. doi: 10.1016/j.bpj.2012.03.045
Li, Y., Kranias, E. G., Mignery, G. A., and Bers, D. M. (2002). Protein kinase A phosphorylation of the ryanodine receptor does not affect calcium sparks in mouse ventricular myocytes. Circ. Res. 90, 309–316. doi: 10.1161/hh0302.105660
Li, Z. M., Chen, L. X., and Li, H. (2019). Voltage-gated Sodium Channels and Blockers: An Overview and Where Will They Go? Curr. Med. Sci. 39, 863–873. doi: 10.1007/s11596-019-2117-0
Liao, Y., Asakura, M., Takashima, S., Kato, H., Asano, Y., Shintani, Y., et al. (2005). Amlodipine ameliorates myocardial hypertrophy by inhibiting EGFR phosphorylation. Biochem. Biophys. Res. Commun. 327, 1083–1087. doi: 10.1016/j.bbrc.2004.12.112
Ling, H., Zhang, T., Pereira, L., Means, C. K., Cheng, H., Gu, Y., et al. (2009). Requirement for Ca2+/calmodulin-dependent kinase II in the transition from pressure overload-induced cardiac hypertrophy to heart failure in mice. J. Clin. Invest. 119, 1230–1240. doi: 10.1172/jci38022
Lipskaia, L., Keuylian, Z., Blirando, K., Mougenot, N., Jacquet, A., Rouxel, C., et al. (2014). Expression of sarco (endo) plasmic reticulum calcium ATPase (SERCA) system in normal mouse cardiovascular tissues, heart failure and atherosclerosis. Biochim. Biophys. Acta 1843, 2705–2718. doi: 10.1016/j.bbamcr.2014.08.002
Little, R., Cartwright, E. J., Neyses, L., and Austin, C. (2016). Plasma membrane calcium ATPases (PMCAs) as potential targets for the treatment of essential hypertension. Pharmacol. Therapeut. 159, 23–34. doi: 10.1016/j.pharmthera.2016.01.013
Little, R., Zi, M., Hammad, S. K., Nguyen, L., Njegic, A., Kurusamy, S., et al. (2017). Reduced expression of PMCA1 is associated with increased blood pressure with age which is preceded by remodelling of resistance arteries. Aging Cell 16, 1104–1113. doi: 10.1111/acel.12637
Liu, H., Chatel, S., Simard, C., Syam, N., Salle, L., Probst, V., et al. (2013). Molecular genetics and functional anomalies in a series of 248 Brugada cases with 11 mutations in the TRPM4 channel. PLoS One 8:e54131. doi: 10.1371/journal.pone.0054131
Liu, N., Ruan, Y., Denegri, M., Bachetti, T., Li, Y., Colombi, B., et al. (2011). Calmodulin kinase II inhibition prevents arrhythmias in RyR2(R4496C+/-) mice with catecholaminergic polymorphic ventricular tachycardia. J. Mol. Cell Cardiol. 50, 214–222. doi: 10.1016/j.yjmcc.2010.10.001
Lohse, M. J., Engelhardt, S., and Eschenhagen, T. (2003). What is the role of beta-adrenergic signaling in heart failure? Circ. Res. 93, 896–906. doi: 10.1161/01.res.0000102042.83024.ca
Lorenz, K., Schmitt, J. P., Vidal, M., and Lohse, M. J. (2009). Cardiac hypertrophy: targeting Raf/MEK/ERK1/2-signaling. Int. J. Biochem. Cell Biol. 41, 2351–2355. doi: 10.1016/j.biocel.2009.08.002
Lou, Q., Janardhan, A., and Efimov, I. R. (2012). Remodeling of calcium handling in human heart failure. Adv. Exp. Med. Biol. 740, 1145–1174. doi: 10.1007/978-94-007-2888-2_52
Louch, W. E., Hougen, K., Mork, H. K., Swift, F., Aronsen, J. M., Sjaastad, I., et al. (2010). Sodium accumulation promotes diastolic dysfunction in end-stage heart failure following Serca2 knockout. J. Physiol. 588, 465–478. doi: 10.1113/jphysiol.2009.183517
Luo, W., Grupp, I. L., Harrer, J., Ponniah, S., Grupp, G., Duffy, J. J., et al. (1994). Targeted ablation of the phospholamban gene is associated with markedly enhanced myocardial contractility and loss of beta-agonist stimulation. Circ. Res. 75, 401–409. doi: 10.1161/01.res.75.3.401
Luongo, T. S., Lambert, J. P., Yuan, A., Zhang, X., Gross, P., Song, J., et al. (2015). The Mitochondrial Calcium Uniporter Matches Energetic Supply with Cardiac Workload during Stress and Modulates Permeability Transition. Cell Rep. 12, 23–34. doi: 10.1016/j.celrep.2015.06.017
Lymperopoulos, A., Rengo, G., and Koch, W. J. (2013). Adrenergic nervous system in heart failure: pathophysiology and therapy. Circ. Res. 113, 739–753. doi: 10.1161/circresaha.113.300308
Lyon, A. R., Bannister, M. L., Collins, T., Pearce, E., Sepehripour, A. H., Dubb, S. S., et al. (2011). SERCA2a gene transfer decreases sarcoplasmic reticulum calcium leak and reduces ventricular arrhythmias in a model of chronic heart failure. Circ. Arrhythm. Electrophysiol. 4, 362–372. doi: 10.1161/circep.110.961615
Madrid, A. H., Bueno, M. G., Rebollo, J. M., Marin, I., Pena, G., Bernal, E., et al. (2002). Use of irbesartan to maintain sinus rhythm in patients with long-lasting persistent atrial fibrillation: a prospective and randomized study. Circulation 106, 331–336. doi: 10.1161/01.cir.0000022665.18619.83
Maggioni, A. P., Anand, I., Gottlieb, S. O., Latini, R., Tognoni, G., Cohn, J. N., et al. (2002). Effects of valsartan on morbidity and mortality in patients with heart failure not receiving angiotensin-converting enzyme inhibitors. J. Am. Coll. Cardiol. 40, 1414–1421. doi: 10.1016/s0735-1097(02)02304-5
Makarewich, C. A., Correll, R. N., Gao, H., Zhang, H., Yang, B., Berretta, R. M., et al. (2012). A caveolae-targeted L-type Ca(2)+ channel antagonist inhibits hypertrophic signaling without reducing cardiac contractility. Circ. Res. 110, 669–674. doi: 10.1161/circresaha.111.264028
Mangoni, M. E., Traboulsie, A., Leoni, A. L., Couette, B., Marger, L., Le Quang, K., et al. (2006). Bradycardia and slowing of the atrioventricular conduction in mice lacking CaV3.1/alpha1G T-type calcium channels. Circ. Res. 98, 1422–1430. doi: 10.1161/01.res.0000225862.14314.49
Marx, S. O., Reiken, S., Hisamatsu, Y., Jayaraman, T., Burkhoff, D., Rosemblit, N., et al. (2000). PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell 101, 365–376. doi: 10.1016/s0092-8674(00)80847-8
Masarone, D., Limongelli, G., Rubino, M., Valente, F., Vastarella, R., Ammendola, E., et al. (2017). Management of arrhythmias in heart failure. J. Cardiovasc. Dev. Dis. 4:3. doi: 10.1007/978-3-642-75326-8_1
Mathar, I., Kecskes, M., Van der Mieren, G., Jacobs, G., Camacho Londono, J. E., Uhl, S., et al. (2014). Increased beta-adrenergic inotropy in ventricular myocardium from Trpm4-/- mice. Circ. Res. 114, 283–294. doi: 10.1161/circresaha.114.302835
Mattiazzi, A., and Kranias, E. G. (2014). The role of CaMKII regulation of phospholamban activity in heart disease. Front. Pharmacol. 5:5. doi: 10.3389/fphar.2014.00005
Mattiazzi, A., Mundina-Weilenmann, C., Guoxiang, C., Vittone, L., and Kranias, E. (2005). Role of phospholamban phosphorylation on Thr17 in cardiac physiological and pathological conditions. Cardiovasc. Res. 68, 366–375. doi: 10.1016/j.cardiores.2005.08.010
Mattiello, J. A., Margulies, K. B., Jeevanandam, V., and Houser, S. R. (1998). Contribution of reverse-mode sodium-calcium exchange to contractions in failing human left ventricular myocytes. Cardiovasc. Res. 37, 424–431. doi: 10.1016/s0008-6363(97)00271-x
Maxwell, J. T., Domeier, T. L., and Blatter, L. A. (2012). Dantrolene prevents arrhythmogenic Ca2+ release in heart failure. Am. J. Physiol. Heart Circ. Physiol. 302, H953–H963.
Mazur, A., Roden, D. M., and Anderson, M. E. (1999). Systemic administration of calmodulin antagonist W-7 or protein kinase A inhibitor H-8 prevents torsade de pointes in rabbits. Circulation 100, 2437–2442. doi: 10.1161/01.cir.100.24.2437
McCauley, M. D., and Wehrens, X. H. (2011). Targeting ryanodine receptors for anti-arrhythmic therapy. Acta Pharmacol. Sin. 32, 749–757. doi: 10.1038/aps.2011.44
McCauley, M., Vallabhajosyula, S., and Darbar, D. (2016). Proarrhythmic and torsadogenic effects of potassium channel blockers in patients. Card Electrophysiol. Clin. 8, 481–493. doi: 10.1016/j.ccep.2016.02.009
McMurray, J. J. (2004). Angiotensin inhibition in heart failure. J. Renin. Angiotensin. Aldosterone Syst. 5(Suppl. 1), S17–S22.
McMurray, J. J., Ostergren, J., Swedberg, K., Granger, C. B., Held, P., Michelson, E. L., et al. (2003). Effects of candesartan in patients with chronic heart failure and reduced left-ventricular systolic function taking angiotensin-converting-enzyme inhibitors: the CHARM-Added trial. Lancet 362, 767–771. doi: 10.1016/s0140-6736(03)14283-3
Menick, D. R., Renaud, L., Buchholz, A., Muller, J. G., Zhou, H., Kappler, C. S., et al. (2007). Regulation of Ncx1 gene expression in the normal and hypertrophic heart. Ann. N. Y. Acad. Sci. 1099, 195–203. doi: 10.1196/annals.1387.058
Mercadier, J. J., Lompre, A. M., Duc, P., Boheler, K. R., Fraysse, J. B., Wisnewsky, C., et al. (1990). Altered sarcoplasmic reticulum Ca2(+)-ATPase gene expression in the human ventricle during end-stage heart failure. J. Clin. Invest. 85, 305–309. doi: 10.1172/jci114429
Micheletti, R., Mattera, G. G., Rocchetti, M., Schiavone, A., Loi, M. F., Zaza, A., et al. (2002). Pharmacological profile of the novel inotropic agent (E,Z)-3-((2-aminoethoxy)imino)androstane-6,17-dione hydrochloride (PST2744). J. Pharmacol. Exp. Ther. 303, 592–600. doi: 10.1124/jpet.102.038331
Micheletti, R., Palazzo, F., Barassi, P., Giacalone, G., Ferrandi, M., Schiavone, A., et al. (2007). Istaroxime, a stimulator of sarcoplasmic reticulum calcium adenosine triphosphatase isoform 2a activity, as a novel therapeutic approach to heart failure. Am. J. Cardiol. 99, 24A–32A.
Miyamoto, S., Zhu, B. M., Kamiya, K., Nagasawa, Y., and Hashimoto, K. (2002). KB-R7943, a Na+/Ca2+ exchange inhibitor, does not suppress ischemia/reperfusion arrhythmias nor digitalis arrhythmias in dogs. Jpn. J. Pharmacol. 90, 229–235. doi: 10.1254/jjp.90.229
Mohamed, T. M. A., Abou-Leisa, R., Stafford, N., Maqsood, A., Zi, M., Prehar, S., et al. (2016). The plasma membrane calcium ATPase 4 signalling in cardiac fibroblasts mediates cardiomyocyte hypertrophy. Nat. Commun. 7:11074.
Mohamed, T. M., Abou-Leisa, R., Baudoin, F., Stafford, N., Neyses, L., Cartwright, E. J., et al. (2013a). Development and characterization of a novel fluorescent indicator protein PMCA4-GCaMP2 in cardiomyocytes. J. Mol. Cell Cardiol. 63, 57–68. doi: 10.1016/j.yjmcc.2013.07.007
Mohamed, T. M., Zakeri, S. A., Baudoin, F., Wolf, M., Oceandy, D., Cartwright, E. J., et al. (2013b). Optimisation and validation of a high throughput screening compatible assay to identify inhibitors of the plasma membrane calcium ATPase pump–a novel therapeutic target for contraception and malaria. J. Pharm. Pharm. Sci. 16, 217–230.
Monfredi, O., Dobrzynski, H., Mondal, T., Boyett, M. R., and Morris, G. M. (2010). The anatomy and physiology of the sinoatrial node–a contemporary review. Pacing Clin. Electrophysiol. 33, 1392–1406. doi: 10.1111/j.1540-8159.2010.02838.x
Morine, K. J., Paruchuri, V., Qiao, X., Aronovitz, M., Huggins, G. S., DeNofrio, D., et al. (2016). Endoglin selectively modulates transient receptor potential channel expression in left and right heart failure. Cardiovasc. Pathol. 25, 478–482. doi: 10.1016/j.carpath.2016.08.004
Most, P., Pleger, S. T., Volkers, M., Heidt, B., Boerries, M., Weichenhan, D., et al. (2004). Cardiac adenoviral S100A1 gene delivery rescues failing myocardium. J. Clin. Invest. 114, 1550–1563. doi: 10.1172/jci21454
Most, P., Seifert, H., Gao, E., Funakoshi, H., Volkers, M., Heierhorst, J., et al. (2006). Cardiac S100A1 protein levels determine contractile performance and propensity toward heart failure after myocardial infarction. Circulation 114, 1258–1268. doi: 10.1161/circulationaha.106.622415
Mukai, M., Terada, H., Sugiyama, S., Satoh, H., and Hayashi, H. (2000). Effects of a selective inhibitor of Na+/Ca2+ exchange, KB-R7943, on reoxygenation-induced injuries in guinea pig papillary muscles. J. Cardiovasc. Pharmacol. 35, 121–128. doi: 10.1097/00005344-200001000-00016
Munch, G., Bolck, B., Karczewski, P., and Schwinger, R. H. (2002). Evidence for calcineurin-mediated regulation of SERCA 2a activity in human myocardium. J. Mol. Cell Cardiol. 34, 321–334. doi: 10.1006/jmcc.2001.1515
Mustroph, J., Neef, S., and Maier, L. S. (2017). CaMKII as a target for arrhythmia suppression. Pharmacol. Ther. 176, 22–31. doi: 10.1016/j.pharmthera.2016.10.006
Nagai, R., Zarain-Herzberg, A., Brandl, C. J., Fujii, J., Tada, M., MacLennan, D. H., et al. (1989). Regulation of myocardial Ca2+-ATPase and phospholamban mRNA expression in response to pressure overload and thyroid hormone. Proc. Natl. Acad. Sci. U.S.A. 86, 2966–2970. doi: 10.1073/pnas.86.8.2966
Nagasawa, Y., Zhu, B. M., Chen, J., Kamiya, K., Miyamoto, S., and Hashimoto, K. (2005). Effects of SEA0400, a Na+/Ca2+ exchange inhibitor, on ventricular arrhythmias in the in vivo dogs. Eur. J. Pharmacol. 506, 249–255. doi: 10.1016/j.ejphar.2004.11.011
Nakamura, M., and Sadoshima, J. (2018). Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 15, 387–407. doi: 10.1038/s41569-018-0007-y
Neumann, J., Eschenhagen, T., Jones, L. R., Linck, B., Schmitz, W., Scholz, H., et al. (1997). Increased expression of cardiac phosphatases in patients with end-stage heart failure. J. Mol. Cell Cardiol. 29, 265–272. doi: 10.1006/jmcc.1996.0271
Nilius, B., Prenen, J., Droogmans, G., Voets, T., Vennekens, R., Freichel, M., et al. (2003). Voltage dependence of the Ca2+-activated cation channel TRPM4. J. Biol. Chem. 278, 30813–30820. doi: 10.1074/jbc.m305127200
Nilius, B., Prenen, J., Tang, J., Wang, C., Owsianik, G., Janssens, A., et al. (2005). Regulation of the Ca2+ sensitivity of the nonselective cation channel TRPM4. J. Biol. Chem. 280, 6423–6433. doi: 10.1074/jbc.m411089200
Oceandy, D., Cartwright, E. J., Emerson, M., Prehar, S., Baudoin, F. M., Zi, M., et al. (2007). Neuronal nitric oxide synthase signaling in the heart is regulated by the sarcolemmal calcium pump 4b. Circulation 115, 483–492. doi: 10.1161/circulationaha.106.643791
Odnoshivkina, U. G., Sytchev, V. I., Nurullin, L. F., Giniatullin, A. R., Zefirov, A. L., and Petrov, A. M. (2015). beta2-adrenoceptor agonist-evoked reactive oxygen species generation in mouse atria: implication in delayed inotropic effect. Eur. J. Pharmacol. 765, 140–153. doi: 10.1016/j.ejphar.2015.08.020
Ohba, T., Watanabe, H., Murakami, M., Takahashi, Y., Iino, K., Kuromitsu, S., et al. (2007). Upregulation of TRPC1 in the development of cardiac hypertrophy. J. Mol. Cell Cardiol. 42, 498–507. doi: 10.1016/j.yjmcc.2006.10.020
Okuyama, Y., Hirawa, N., Fujita, M., Fujiwara, A., Ehara, Y., Yatsu, K., et al. (2018). The effects of anti-hypertensive drugs and the mechanism of hypertension in vascular smooth muscle cell-specific ATP2B1 knockout mice. Hypertens. Res. 41, 80–87. doi: 10.1038/hr.2017.92
Ortega, A., Rosello-Lleti, E., Tarazon, E., Gil-Cayuela, C., Lago, F., Gonzalez-Juanatey, J. R., et al. (2016). TRPM7 is down-regulated in both left atria and left ventricle of ischaemic cardiomyopathy patients and highly related to changes in ventricular function. ESC Heart Fail. 3, 220–224. doi: 10.1002/ehf2.12085
Paavola, J., Schliffke, S., Rossetti, S., Kuo, I. Y., Yuan, S., Sun, Z., et al. (2013). Polycystin-2 mutations lead to impaired calcium cycling in the heart and predispose to dilated cardiomyopathy. J. Mol. Cell Cardiol. 58, 199–208. doi: 10.1016/j.yjmcc.2013.01.015
Packer, M., Fowler, M. B., Roecker, E. B., Coats, A. J., Katus, H. A., Krum, H., et al. (2002). Effect of carvedilol on the morbidity of patients with severe chronic heart failure: results of the carvedilol prospective randomized cumulative survival (COPERNICUS) study. Circulation 106, 2194–2199. doi: 10.1161/01.cir.0000035653.72855.bf
Packer, M., O’Connor, C. M., Ghali, J. K., Pressler, M. L., Carson, P. E., Belkin, R. N., et al. (1996). Effect of amlodipine on morbidity and mortality in severe chronic heart failure. Prospective Randomized Amlodipine Survival Evaluation Study Group. N. Engl. J. Med. 335, 1107–1114.
Pande, J., Mallhi, K. K., Sawh, A., Szewczyk, M. M., Simpson, F., and Grover, A. K. (2006). Aortic smooth muscle and endothelial plasma membrane Ca2+ pump isoforms are inhibited differently by the extracellular inhibitor caloxin 1b1. Am. J. Physiol. Cell Physiol. 290, C1341–C1349.
Pande, J., Szewczyk, M. M., and Grover, A. K. (2011). Allosteric inhibitors of plasma membrane Ca pumps: Invention and applications of caloxins. World J. Biol. Chem. 2, 39–47.
Paolocci, N., Saavedra, W. F., Miranda, K. M., Martignani, C., Isoda, T., Hare, J. M., et al. (2001). Nitroxyl anion exerts redox-sensitive positive cardiac inotropy in vivo by calcitonin gene-related peptide signaling. Proc. Natl. Acad. Sci. U.S.A. 98, 10463–10468. doi: 10.1073/pnas.181191198
Patel, K., Fonarow, G. C., Ahmed, M., Morgan, C., Kilgore, M., Love, T. E., et al. (2014). Calcium channel blockers and outcomes in older patients with heart failure and preserved ejection fraction. Circ. Heart Fail. 7, 945–952. doi: 10.1161/circheartfailure.114.001301
Pedersen, O. D., Bagger, H., Kober, L., and Torp-Pedersen, C. (1999). Trandolapril reduces the incidence of atrial fibrillation after acute myocardial infarction in patients with left ventricular dysfunction. Circulation 100, 376–380. doi: 10.1161/01.cir.100.4.376
Penny, W. F., Henry, T. D., Watkins, M. W., Patel, A. N., and Hammond, H. K. (2018). Design of a Phase 3 trial of intracoronary administration of human adenovirus 5 encoding human adenylyl cyclase type 6 (RT-100) gene transfer in patients with heart failure with reduced left ventricular ejection fraction: The FLOURISH Clinical Trial. Am. Heart J. 201, 111–116. doi: 10.1016/j.ahj.2018.04.005
Penttinen, K., Swan, H., Vanninen, S., Paavola, J., Lahtinen, A. M., Kontula, K., et al. (2015). Antiarrhythmic effects of dantrolene in patients with catecholaminergic polymorphic ventricular tachycardia and replication of the responses using iPSC Models. PLoS One 10:e0125366. doi: 10.1371/journal.pone.0125366
Periasamy, M., Reed, T. D., Liu, L. H., Ji, Y., Loukianov, E., Paul, R. J., et al. (1999). Impaired cardiac performance in heterozygous mice with a null mutation in the sarco(endo)plasmic reticulum Ca2+-ATPase isoform 2 (SERCA2) gene. J. Biol. Chem. 274, 2556–2562. doi: 10.1074/jbc.274.4.2556
Pinilla-Vera, M., Hahn, V. S., and Kass, D. A. (2019). Leveraging signaling pathways to treat heart failure with reduced ejection fraction. Circ. Res. 124, 1618–1632. doi: 10.1161/circresaha.119.313682
Pinto, A. R., Ilinykh, A., Ivey, M. J., Kuwabara, J. T., D’Antoni, M. L., Debuque, R., et al. (2016). Revisiting cardiac cellular composition. Circ. Res. 118, 400–409. doi: 10.1161/circresaha.115.307778
Pinton, P., Giorgi, C., Siviero, R., Zecchini, E., and Rizzuto, R. (2008). Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 27, 6407–6418. doi: 10.1038/onc.2008.308
Pinz, I., Tian, R., Belke, D., Swanson, E., Dillmann, W., and Ingwall, J. S. (2011). Compromised myocardial energetics in hypertrophied mouse hearts diminish the beneficial effect of overexpressing SERCA2a. J. Biol. Chem. 286, 10163–10168. doi: 10.1074/jbc.m110.210757
Piper, H. M., Balser, C., Ladilov, Y. V., Schafer, M., Siegmund, B., Ruiz-Meana, M., et al. (1996). The role of Na+/H+ exchange in ischemia-reperfusion. Basic Res. Cardiol. 91, 191–202.
Pitt, B., Pfeffer, M. A., Assmann, S. F., Boineau, R., Anand, I. S., Claggett, B., et al. (2014). Spironolactone for heart failure with preserved ejection fraction. N. Engl. J. Med. 370, 1383–1392.
Pitt, B., Remme, W., Zannad, F., Neaton, J., Martinez, F., Roniker, B., et al. (2003). Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N. Engl. J. Med. 348, 1309–1321. doi: 10.1056/nejmoa030207
Pleger, S. T., Most, P., Boucher, M., Soltys, S., Chuprun, J. K., Pleger, W., et al. (2007). Stable myocardial-specific AAV6-S100A1 gene therapy results in chronic functional heart failure rescue. Circulation 115, 2506–2515. doi: 10.1161/circulationaha.106.671701
Pleger, S. T., Remppis, A., Heidt, B., Volkers, M., Chuprun, J. K., Kuhn, M., et al. (2005). S100A1 gene therapy preserves in vivo cardiac function after myocardial infarction. Mol. Ther. 12, 1120–1129. doi: 10.1016/j.ymthe.2005.08.002
Pleger, S. T., Shan, C., Ksienzyk, J., Bekeredjian, R., Boekstegers, P., Hinkel, R., et al. (2011). Cardiac AAV9-S100A1 gene therapy rescues post-ischemic heart failure in a preclinical large animal model. Sci. Transl. Med. 3:92ra64. doi: 10.1126/scitranslmed.3002097
Pogwizd, S. M., Schlotthauer, K., Li, L., Yuan, W., and Bers, D. M. (2001). Arrhythmogenesis and contractile dysfunction in heart failure: Roles of sodium-calcium exchange, inward rectifier potassium current, and residual beta-adrenergic responsiveness. Circ. Res. 88, 1159–1167. doi: 10.1161/hh1101.091193
Ponikowski, P., Voors, A. A., Anker, S. D., Bueno, H., Cleland, J. G., Coats, A. J., et al. (2016). 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. J. Heart Fail. 18, 891–975.
Port, J. D., and Bristow, M. R. (2001). Altered beta-adrenergic receptor gene regulation and signaling in chronic heart failure. J. Mol. Cell Cardiol. 33, 887–905. doi: 10.1006/jmcc.2001.1358
Pu, J., Zhang, C. T., Bai, R., Liu, N., Li, Y., and Wang, L. (2005). [Calmodulin antagonist inhibits torsade de pointes induced by d-sotalol in an isolated rabbit heart model]. Zhonghua Xin Xue Guan Bing Za Zhi. 33, 364–368.
Qi, Z., Wong, C. K., Suen, C. H., Wang, J., Long, C., Sauer, H., et al. (2016). TRPC3 regulates the automaticity of embryonic stem cell-derived cardiomyocytes. Int. J. Cardiol. 203, 169–181. doi: 10.1016/j.ijcard.2015.10.018
Ramza, B. M., Tan, R. C., Osaka, T., and Joyner, R. W. (1990). Cellular mechanism of the functional refractory period in ventricular muscle. Circ. Res. 66, 147–162. doi: 10.1161/01.res.66.1.147
Rastogi, A., Novak, E., Platts, A. E., and Mann, D. L. (2017). Epidemiology, pathophysiology and clinical outcomes for heart failure patients with a mid-range ejection fraction. Eur. J. Heart Fail 19, 1597–1605. doi: 10.1002/ejhf.879
Reiken, S., Gaburjakova, M., Gaburjakova, J., He, K. L., Prieto, A., Becker, E., et al. (2001). beta-adrenergic receptor blockers restore cardiac calcium release channel (ryanodine receptor) structure and function in heart failure. Circulation 104, 2843–2848. doi: 10.1161/hc4701.099578
Reiken, S., Wehrens, X. H., Vest, J. A., Barbone, A., Klotz, S., Mancini, D., et al. (2003). Beta-blockers restore calcium release channel function and improve cardiac muscle performance in human heart failure. Circulation 107, 2459–2466. doi: 10.1161/01.cir.0000068316.53218.49
Respress, J. L., van Oort, R. J., Li, N., Rolim, N., Dixit, S. S., deAlmeida, A., et al. (2012). Role of RyR2 phosphorylation at S2814 during heart failure progression. Circ. Res. 110, 1474–1483. doi: 10.1161/circresaha.112.268094
Richard, S., Leclercq, F., Lemaire, S., Piot, C., and Nargeot, J. (1998). Ca2+ currents in compensated hypertrophy and heart failure. Cardiovasc. Res. 37, 300–311. doi: 10.1016/s0008-6363(97)00273-3
Rios, F. J., Zou, Z. G., Harvey, A. P., Harvey, K. Y., Nosalski, R., Anyfanti, P., et al. (2020). Chanzyme TRPM7 protects against cardiovascular inflammation and fibrosis. Cardiovasc. Res. 116, 721–735. doi: 10.1093/cvr/cvz164
Ritterhoff, J., and Most, P. (2012). Targeting S100A1 in heart failure. Gene Ther. 19, 613–621. doi: 10.1038/gt.2012.8
Roe, A. T., Frisk, M., and Louch, W. E. (2015). Targeting cardiomyocyte Ca2+ homeostasis in heart failure. Curr. Pharm. Des. 21, 431–448. doi: 10.2174/138161282104141204124129
Rohrbach, S., Niemann, B., Silber, R. E., and Holtz, J. (2005). Neuregulin receptors erbB2 and erbB4 in failing human myocardium – depressed expression and attenuated activation. Basic Res Cardiol. 100, 240–249. doi: 10.1007/s00395-005-0514-4
Rohrbach, S., Yan, X., Weinberg, E. O., Hasan, F., Bartunek, J., Marchionni, M. A., et al. (1999). Neuregulin in cardiac hypertrophy in rats with aortic stenosis. Differential expression of erbB2 and erbB4 receptors. Circulation 100, 407–412. doi: 10.1161/01.cir.100.4.407
Rose, B. A., Force, T., and Wang, Y. (2010). Mitogen-activated protein kinase signaling in the heart: angels versus demons in a heart-breaking tale. Physiol. Rev. 90, 1507–1546. doi: 10.1152/physrev.00054.2009
Rosso, R., Kalman, J. M., Rogowski, O., Diamant, S., Birger, A., Biner, S., et al. (2007). Calcium channel blockers and beta-blockers versus beta-blockers alone for preventing exercise-induced arrhythmias in catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 4, 1149–1154. doi: 10.1016/j.hrthm.2007.05.017
Roth, G. A., Johnson, C., Abajobir, A., Abd-Allah, F., Abera, S. F., Abyu, G., et al. (2017). Global, Regional, and National Burden of Cardiovascular Diseases for 10 Causes, 1990 to 2015. J. Am. Coll. Cardiol. 70, 1–25.
Ruiz-Meana, M., Minguet, M., Bou-Teen, D., Miro-Casas, E., Castans, C., Castellano, J., et al. (2019). Ryanodine receptor glycation favors mitochondrial damage in the senescent heart. Circulation 139, 949–964. doi: 10.1161/circulationaha.118.035869
Sabbah, H. N., Imai, M., Cowart, D., Amato, A., Carminati, P., and Gheorghiade, M. (2007). Hemodynamic properties of a new-generation positive luso-inotropic agent for the acute treatment of advanced heart failure. Am. J. Cardiol. 99, 41A–46A.
Sah, R., Mesirca, P., Mason, X., Gibson, W., Bates-Withers, C., Van den Boogert, M., et al. (2013a). Timing of myocardial trpm7 deletion during cardiogenesis variably disrupts adult ventricular function, conduction, and repolarization. Circulation 128, 101–114. doi: 10.1161/circulationaha.112.000768
Sah, R., Mesirca, P., Van den Boogert, M., Rosen, J., Mably, J., Mangoni, M. E., et al. (2013b). Ion channel-kinase TRPM7 is required for maintaining cardiac automaticity. Proc. Natl. Acad. Sci. U.S.A. 110, E3037–E3046.
Samuel, T. J., Rosenberry, R. P., Lee, S., and Pan, Z. (2018). Correcting calcium dysregulation in chronic heart failure using SERCA2a gene therapy. Int. J. Mol. Sci. 19:1086. doi: 10.3390/ijms19041086
Sanchez-Alonso, J. L., Bhargava, A., O’Hara, T., Glukhov, A. V., Schobesberger, S., Bhogal, N., et al. (2016). Microdomain-Specific Modulation of L-type calcium channels leads to triggered ventricular arrhythmia in heart failure. Circ. Res. 119, 944–955. doi: 10.1161/circresaha.116.308698
Sasaki, K., Makiyama, T., Yoshida, Y., Wuriyanghai, Y., Kamakura, T., Nishiuchi, S., et al. (2016). Patient-Specific Human Induced Pluripotent Stem Cell Model Assessed with Electrical Pacing Validates S107 as a potential therapeutic agent for catecholaminergic polymorphic ventricular tachycardia. PLoS One 11:e0164795. doi: 10.1371/journal.pone.0164795
Satoh, S., Tanaka, H., Ueda, Y., Oyama, J., Sugano, M., Sumimoto, H., et al. (2007). Transient receptor potential (TRP) protein 7 acts as a G protein-activated Ca2+ channel mediating angiotensin II-induced myocardial apoptosis. Mol. Cell Biochem. 294, 205–215. doi: 10.1007/s11010-006-9261-0
Sawada, K. I., and Kawamura, K. (1991). Architecture of myocardial cells in human cardiac ventricles with concentric and eccentric hypertrophy as demonstrated by quantitative scanning electron microscopy. Heart Vessels 6, 129–142. doi: 10.1007/bf02058278
Schillinger, W., Janssen, P. M., Emami, S., Henderson, S. A., Ross, R. S., Teucher, N., et al. (2000). Impaired contractile performance of cultured rabbit ventricular myocytes after adenoviral gene transfer of Na(+)-Ca(2+) exchanger. Circ. Res. 87, 581–587. doi: 10.1161/01.res.87.7.581
Schroder, F., Handrock, R., Beuckelmann, D. J., Hirt, S., Hullin, R., Priebe, L., et al. (1998). Increased availability and open probability of single L-type calcium channels from failing compared with nonfailing human ventricle. Circulation 98, 969–976. doi: 10.1161/01.cir.98.10.969
Schwinger, R. H., Bohm, M., Schmidt, U., Karczewski, P., Bavendiek, U., Flesch, M., et al. (1995). Unchanged protein levels of SERCA II and phospholamban but reduced Ca2+ uptake and Ca(2+)-ATPase activity of cardiac sarcoplasmic reticulum from dilated cardiomyopathy patients compared with patients with nonfailing hearts. Circulation 92, 3220–3228. doi: 10.1161/01.cir.92.11.3220
Seo, K., Rainer, P. P., Shalkey Hahn, V., Lee, D. I., Jo, S. H., Andersen, A., et al. (2014). Combined TRPC3 and TRPC6 blockade by selective small-molecule or genetic deletion inhibits pathological cardiac hypertrophy. Proc. Natl. Acad. Sci. U.S.A. 111, 1551–1556. doi: 10.1073/pnas.1308963111
Seth, M., Zhang, Z. S., Mao, L., Graham, V., Burch, J., Stiber, J., et al. (2009). TRPC1 channels are critical for hypertrophic signaling in the heart. Circ. Res. 105, 1023–1030. doi: 10.1161/circresaha.109.206581
Shah, S. J., Blair, J. E., Filippatos, G. S., Macarie, C., Ruzyllo, W., Korewicki, J., et al. (2009). Effects of istaroxime on diastolic stiffness in acute heart failure syndromes: results from the Hemodynamic, Echocardiographic, and Neurohormonal Effects of Istaroxime, a Novel Intravenous Inotropic and Lusitropic Agent: a Randomized Controlled Trial in Patients Hospitalized with Heart Failure (HORIZON-HF) trial. Am. Heart J. 157, 1035–1041. doi: 10.1016/j.ahj.2009.03.007
Shan, J., Betzenhauser, M. J., Kushnir, A., Reiken, S., Meli, A. C., Wronska, A., et al. (2010). Role of chronic ryanodine receptor phosphorylation in heart failure and beta-adrenergic receptor blockade in mice. J. Clin. Invest. 120, 4375–4387. doi: 10.1172/jci37649
Simard, C., Hof, T., Keddache, Z., Launay, P., and Guinamard, R. (2013). The TRPM4 non-selective cation channel contributes to the mammalian atrial action potential. J. Mol. Cell Cardiol. 59, 11–19. doi: 10.1016/j.yjmcc.2013.01.019
Sombie, H. K., Kologo, J. K., Tchelougou, D., Ouedraogo, S. Y., Ouattara, A. K., Compaore, T. R., et al. (2019). Positive association between ATP2B1 rs17249754 and essential hypertension: a case-control study in Burkina Faso. West Africa. BMC Cardiovasc. Disord. 19:155. doi: 10.1186/s12872-019-1136-x
Stafford, N., Wilson, C., Oceandy, D., Neyses, L., and Cartwright, E. J. (2017). The Plasma Membrane Calcium ATPases and Their Role as Major New Players in Human Disease. Physiol. Rev. 97, 1089–1125. doi: 10.1152/physrev.00028.2016
Stallmeyer, B., Zumhagen, S., Denjoy, I., Duthoit, G., Hebert, J. L., Ferrer, X., et al. (2012). Mutational spectrum in the Ca(2+)–activated cation channel gene TRPM4 in patients with cardiac conductance disturbances. Hum. Mutat. 33, 109–117. doi: 10.1002/humu.21599
Sunggip, C., Shimoda, K., Oda, S., Tanaka, T., Nishiyama, K., Mangmool, S., et al. (2018). TRPC5-eNOS axis negatively regulates ATP-induced cardiomyocyte hypertrophy. Front. Pharmacol. 9:523. doi: 10.3389/fphar.2018.00523
Szewczyk, M. M., Pande, J., Akolkar, G., and Grover, A. K. (2010). Caloxin 1b3: a novel plasma membrane Ca(2+)-pump isoform 1 selective inhibitor that increases cytosolic Ca(2+) in endothelial cells. Cell Calcium 48, 352–357. doi: 10.1016/j.ceca.2010.10.008
Tabara, Y., Kohara, K., Kita, Y., Hirawa, N., Katsuya, T., Ohkubo, T., et al. (2010). Common variants in the ATP2B1 gene are associated with susceptibility to hypertension: the Japanese Millennium Genome Project. Hypertension 56, 973–980. doi: 10.1161/hypertensionaha.110.153429
Takahashi, K., Takahashi, T., Suzuki, T., Onishi, M., Tanaka, Y., Hamano-Takahashi, A., et al. (2003). Protective effects of SEA0400, a novel and selective inhibitor of the Na+/Ca2+ exchanger, on myocardial ischemia-reperfusion injuries. Eur. J. Pharmacol. 458, 155–162. doi: 10.1016/s0014-2999(02)02732-2
Takeuchi, F., Isono, M., Katsuya, T., Yamamoto, K., Yokota, M., Sugiyama, T., et al. (2010). Blood Pressure and Hypertension Are Associated With 7 Loci in the Japanese Population. Circulation 121, 2302–U140.
Tanaka, H., Nishimaru, K., Aikawa, T., Hirayama, W., Tanaka, Y., and Shigenobu, K. (2002). Effect of SEA0400, a novel inhibitor of sodium-calcium exchanger, on myocardial ionic currents. Br. J. Pharmacol. 135, 1096–1100. doi: 10.1038/sj.bjp.0704574
Tepper, D. (1999). Frontiers in congestive heart failure: Effect of Metoprolol CR/XL in chronic heart failure: Metoprolol CR/XL Randomised Intervention Trial in Congestive Heart Failure (MERIT-HF). Congest. Heart Fail. 5, 184–185.
The Danish Study Group on Verapamil in Myocardial Infarction (1990). Effect of verapamil on mortality and major events after acute myocardial infarction (the Danish Verapamil Infarction Trial II–DAVIT II). Am. J. Cardiol. 66, 779–785 doi: 10.1016/0002-9149(90)90351-z
Thorneloe, K. S., Cheung, M., Bao, W., Alsaid, H., Lenhard, S., Jian, M. Y., et al. (2012). An orally active TRPV4 channel blocker prevents and resolves pulmonary edema induced by heart failure. Sci. Transl. Med. 4:159ra148. doi: 10.1126/scitranslmed.3004276
Tilemann, L., Lee, A., Ishikawa, K., Aguero, J., Rapti, K., Santos-Gallego, C., et al. (2013). SUMO-1 gene transfer improves cardiac function in a large-animal model of heart failure. Sci Transl. Med. 5:211ra159. doi: 10.1126/scitranslmed.3006487
Tita, C., Gilbert, E. M., Van Bakel, A. B., Grzybowski, J., Haas, G. J., Jarrah, M., et al. (2017). A Phase 2a dose-escalation study of the safety, tolerability, pharmacokinetics and haemodynamic effects of BMS-986231 in hospitalized patients with heart failure with reduced ejection fraction. Eur. J. Heart Fail. 19, 1321–1332. doi: 10.1002/ejhf.897
Tzimas, C., Terrovitis, J., Lehnart, S. E., Kranias, E. G., and Sanoudou, D. (2015). Calcium/calmodulin-dependent protein kinase II (CaMKII) inhibition ameliorates arrhythmias elicited by junctin ablation under stress conditions. Heart Rhythm. 12, 1599–1610. doi: 10.1016/j.hrthm.2015.03.043
Ueng, K. C., Tsai, T. P., Yu, W. C., Tsai, C. F., Lin, M. C., Chan, K. C., et al. (2003). Use of enalapril to facilitate sinus rhythm maintenance after external cardioversion of long-standing persistent atrial fibrillation. Results of a prospective and controlled study. Eur. Heart J. 24, 2090–2098. doi: 10.1016/j.ehj.2003.08.014
Venetucci, L. A., Trafford, A. W., O’Neill, S. C., and Eisner, D. A. (2008). The sarcoplasmic reticulum and arrhythmogenic calcium release. Cardiovasc. Res. 77, 285–292. doi: 10.1093/cvr/cvm009
Vest, J. A., Wehrens, X. H., Reiken, S. R., Lehnart, S. E., Dobrev, D., Chandra, P., et al. (2005). Defective cardiac ryanodine receptor regulation during atrial fibrillation. Circulation 111, 2025–2032. doi: 10.1161/01.cir.0000162461.67140.4c
Wachtell, K., Lehto, M., Gerdts, E., Olsen, M. H., Hornestam, B., Dahlof, B., et al. (2005). Angiotensin II receptor blockade reduces new-onset atrial fibrillation and subsequent stroke compared to atenolol: the Losartan Intervention For End Point Reduction in Hypertension (LIFE) study. J. Am. Coll. Cardiol. 45, 712–719. doi: 10.1016/j.jacc.2004.10.068
Wahlquist, C., Jeong, D., Rojas-Munoz, A., Kho, C., Lee, A., Mitsuyama, S., et al. (2014). Inhibition of miR-25 improves cardiac contractility in the failing heart. Nature 508, 531–535. doi: 10.1038/nature13073
Wang, C., Naruse, K., and Takahashi, K. (2018). Role of the TRPM4 channel in cardiovascular physiology and pathophysiology. Cells 7:62. doi: 10.3390/cells7060062
Wang, J., Takahashi, K., Piao, H., Qu, P., and Naruse, K. (2013). 9-Phenanthrol, a TRPM4 inhibitor, protects isolated rat hearts from ischemia-reperfusion injury. PLoS One 8:e70587. doi: 10.1371/journal.pone.0070587
Wang, X., Zhuo, X., Gao, J., Liu, H., Lin, F., and Ma, A. (2019). Neuregulin-1beta partially improves cardiac function in volume-overload heart failure through regulation of abnormal calcium handling. Front. Pharmacol. 10:616. doi: 10.3389/fphar.2019.00616
Wang, Y., Zhang, Y., Li, Y., Zhou, X., Wang, X., Gao, P., et al. (2013). Common variants in the ATP2B1 gene are associated with hypertension and arterial stiffness in Chinese population. Mol. Biol. Rep. 40, 1867–1873. doi: 10.1007/s11033-012-2242-3
Wang, Z., Nolan, B., Kutschke, W., and Hill, J. A. (2001). Na+-Ca2+ exchanger remodeling in pressure overload cardiac hypertrophy. J. Biol. Chem. 276, 17706–17711. doi: 10.1074/jbc.m100544200
Washburn, D. G., Holt, D. A., Dodson, J., McAtee, J. J., Terrell, L. R., Barton, L., et al. (2013). The discovery of potent blockers of the canonical transient receptor channels. TRPC3 and TRPC6, based on an anilino-thiazole pharmacophore. Bioorg. Med. Chem. Lett. 23, 4979–4984. doi: 10.1016/j.bmcl.2013.06.047
Watanabe, H., Chopra, N., Laver, D., Hwang, H. S., Davies, S. S., Roach, D. E., et al. (2009). Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans. Nat. Med. 15, 380–383. doi: 10.1038/nm.1942
Watkins, L. Jr., Burton, J. A., Haber, E., Cant, J. R., Smith, F. W., and Barger, A. C. (1976). The renin-angiotensin-aldosterone system in congestive failure in conscious dogs. J. Clin. Invest. 57, 1606–1617.
Wehrens, X. H., Lehnart, S. E., Reiken, S. R., Deng, S. X., Vest, J. A., Cervantes, D., et al. (2004b). Protection from cardiac arrhythmia through ryanodine receptor-stabilizing protein calstabin2. Science 304, 292–296. doi: 10.1126/science.1094301
Wehrens, X. H., Lehnart, S. E., Reiken, S. R., and Marks, A. R. (2004a). Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ. Res. 94, e61–e70.
Wehrens, X. H., Lehnart, S. E., Reiken, S., Vest, J. A., Wronska, A., and Marks, A. R. (2006). Ryanodine receptor/calcium release channel PKA phosphorylation: a critical mediator of heart failure progression. Proc. Natl. Acad. Sci. U.S.A. 103, 511–518. doi: 10.1073/pnas.0510113103
Weiss, J. N., Garfinkel, A., Karagueuzian, H. S., Chen, P. S., and Qu, Z. (2010). Early afterdepolarizations and cardiac arrhythmias. Heart Rhythm. 7, 1891–1899. doi: 10.1016/j.hrthm.2010.09.017
Weiss, J. N., Nivala, M., Garfinkel, A., and Qu, Z. (2011). Alternans and arrhythmias: from cell to heart. Circ. Res. 108, 98–112. doi: 10.1161/circresaha.110.223586
Wilde, A. A., and Bezzina, C. R. (2005). Genetics of cardiac arrhythmias. Heart 91, 1352–1358. doi: 10.1136/hrt.2004.046334
Wilkins, B. J., Dai, Y. S., Bueno, O. F., Parsons, S. A., Xu, J., Plank, D. M., et al. (2004). Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ. Res. 94, 110–118. doi: 10.1161/01.res.0000109415.17511.18
Wong, M. H., Samal, A. B., Lee, M., Vlach, J., Novikov, N., Niedziela-Majka, A., et al. (2019). The KN-93 Molecule Inhibits Calcium/Calmodulin-Dependent Protein Kinase II (CaMKII) Activity by Binding to Ca(2+)/CaM. J. Mol. Biol. 431, 1440–1459. doi: 10.1016/j.jmb.2019.02.001
Workman, A. J. (2010). Cardiac adrenergic control and atrial fibrillation. Naunyn Schmiedebergs Arch. Pharmacol. 381, 235–249. doi: 10.1007/s00210-009-0474-0
Wu, Q. F., Qian, C., Zhao, N., Dong, Q., Li, J., Wang, B. B., et al. (2017). Activation of transient receptor potential vanilloid 4 involves in hypoxia/reoxygenation injury in cardiomyocytes. Cell Death Dis. 8:e2828. doi: 10.1038/cddis.2017.227
Xie, A., Song, Z., Liu, H., Zhou, A., Shi, G., Wang, Q., et al. (2018). Mitochondrial Ca(2+) influx contributes to arrhythmic risk in nonischemic cardiomyopathy. J Am Heart Assoc. 7:e007805.
Xu, J., Qian, H. X., Hu, S. P., Liu, L. Y., Zhou, M., Feng, M., et al. (2016). Gender-Specific Association of ATP2B1 Variants with Susceptibility to Essential Hypertension in the Han Chinese Population. Biomed. Res. Int. 2016:1910565.
Yamazaki, D., Komazaki, S., Nakanishi, H., Mishima, A., Nishi, M., Yazawa, M., et al. (2009a). Essential role of the TRIC-B channel in Ca2+ handling of alveolar epithelial cells and in perinatal lung maturation. Development 136, 2355–2361. doi: 10.1242/dev.036798
Yamazaki, D., Yamazaki, T., and Takeshima, H. (2009b). New molecular components supporting ryanodine receptor-mediated Ca(2+) release: roles of junctophilin and TRIC channel in embryonic cardiomyocytes. Pharmacol. Ther. 121, 265–272. doi: 10.1016/j.pharmthera.2008.11.004
Yang, H., Hu, M., Guo, J., Ou, X., Cai, T., and Liu, Z. (2016). Pore architecture of TRIC channels and insights into their gating mechanism. Nature 538, 537–541. doi: 10.1038/nature19767
Yazawa, M., Ferrante, C., Feng, J., Mio, K., Ogura, T., Zhang, M., et al. (2007). TRIC channels are essential for Ca2+ handling in intracellular stores. Nature 448, 78–82. doi: 10.1038/nature05928
Yoshida, M., Ma, J., Tomita, T., Morikawa, N., Tanaka, N., Masamura, K., et al. (2005). Mineralocorticoid receptor is overexpressed in cardiomyocytes of patients with congestive heart failure. Congest. Heart Fail. 11, 12–16. doi: 10.1111/j.1527-5299.2005.03722.x
Yoshie, K., Rajendran, P. S., Massoud, L., Mistry, J., Swid, M. A., Wu, X., et al. (2020). Cardiac TRPV1 afferent signaling promotes arrhythmogenic ventricular remodeling after myocardial infarction. JCI Insight 5:e124477.
Yu, Z., Chen, R., Li, M., Yu, Y., Liang, Y., Han, F., et al. (2018). Mitochondrial calcium uniporter inhibition provides cardioprotection in pressure overload-induced heart failure through autophagy enhancement. Int. J. Cardiol. 271, 161–168. doi: 10.1016/j.ijcard.2018.05.054
Zaglia, T., Ceriotti, P., Campo, A., Borile, G., Armani, A., Carullo, P., et al. (2017). Content of mitochondrial calcium uniporter (MCU) in cardiomyocytes is regulated by microRNA-1 in physiologic and pathologic hypertrophy. Proc. Natl. Acad. Sci. U.S.A. 114, E9006–E9015.
Zannad, F., McMurray, J. J., Krum, H., van Veldhuisen, D. J., Swedberg, K., Shi, H., et al. (2011). Eplerenone in patients with systolic heart failure and mild symptoms. N. Engl. J. Med. 364, 11–21.
Zhang, T., Kohlhaas, M., Backs, J., Mishra, S., Phillips, W., Dybkova, N., et al. (2007). CaMKIIdelta isoforms differentially affect calcium handling but similarly regulate HDAC/MEF2 transcriptional responses. J. Biol. Chem. 282, 35078–35087. doi: 10.1074/jbc.m707083200
Zhang, W., Elimban, V., Nijjar, M. S., Gupta, S. K., and Dhalla, N. S. (2003). Role of mitogen-activated protein kinase in cardiac hypertrophy and heart failure. Exp. Clin. Cardiol. 8, 173–183.
Zhang, Y. H., Sun, H. Y., Chen, K. H., Du, X. L., Liu, B., Cheng, L. C., et al. (2012). Evidence for functional expression of TRPM7 channels in human atrial myocytes. Basic Res. Cardiol. 107:282.
Zhao, J., Wang, X., Hang, P., Yun, F., Zhao, H., Xu, W., et al. (2013). Valsartan inhibits transient receptor potential canonical-3 channel in canine atrial fibrillation. Int. J. Cardiol. 168, 4417–4418. doi: 10.1016/j.ijcard.2013.05.029
Zhao, X., Yamazaki, D., Park, K. H., Komazaki, S., Tjondrokoesoemo, A., Nishi, M., et al. (2010). Ca2+ overload and sarcoplasmic reticulum instability in tric-a null skeletal muscle. J. Biol. Chem. 285, 37370–37376.
Zhong, M., Rees, C. M., Terentyev, D., Choi, B. R., Koren, G., and Karma, A. (2018). NCX-mediated subcellular Ca(2+) dynamics underlying early afterdepolarizations in LQT2 cardiomyocytes. Biophys. J. 115, 1019–1032. doi: 10.1016/j.bpj.2018.08.004
Zhou, X., Park, K. H., Yamazaki, D., Lin, P. H., Nishi, M., Ma, Z., et al. (2020). TRIC-A channel maintains store calcium handling by interacting with type 2 ryanodine receptor in cardiac muscle. Circ. Res. 126, 417–435. doi: 10.1161/circresaha.119.316241
Zhu, Y., Qu, J., He, L., Zhang, F., Zhou, Z., Yang, S., et al. (2019). Calcium in vascular smooth muscle cell elasticity and adhesion: novel insights into the mechanism of action. Front. Physiol. 10:852. doi: 10.3389/fphys.2019.00852
Ziaeian, B., and Fonarow, G. C. (2016). Epidemiology and aetiology of heart failure. Nat. Rev. Cardiol. 13, 368–378.
Zsebo, K., Yaroshinsky, A., Rudy, J. J., Wagner, K., Greenberg, B., Jessup, M., et al. (2014). Long-term effects of AAV1/SERCA2a gene transfer in patients with severe heart failure: analysis of recurrent cardiovascular events and mortality. Circ. Res. 114, 101–108. doi: 10.1161/circresaha.113.302421
Keywords: cardiovascular disease, heart failure, arrhythmia, calcium dysregulation, calcium
Citation: Njegic A, Wilson C and Cartwright EJ (2020) Targeting Ca2 + Handling Proteins for the Treatment of Heart Failure and Arrhythmias. Front. Physiol. 11:1068. doi: 10.3389/fphys.2020.01068
Received: 31 May 2020; Accepted: 04 August 2020;
Published: 04 September 2020.
Edited by:
Jun Ren, University of Washington, United StatesReviewed by:
Marisol Ruiz-Meana, University Hospital Vall d’Hebron-Universitat Autònoma de Barcelona, SpainHaixia Xu, Zhongshan Hospital, Fudan University, China
Copyright © 2020 Njegic, Wilson and Cartwright. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elizabeth J. Cartwright, elizabeth.j.cartwright@manchester.ac.uk
†These authors have contributed equally to this work