
Abstract
First-principles calculation reveals that hydrogen, which is abundant in chemical vapor deposition (CVD), can significantly improve the uniformity of nitrogen-vacancy (NV) centers in diamond. It shows that the formation of NV centers can be described as a multi-step process: first, a substitutional N (NC) is preferentially formed at the surface layer over that of either a carbon vacancy (VC) or an in-pane nitrogen-vacancy-hydrogen (NVH) complex. Second, with the help of H, a VC is preferentially incorporated in the newly formed topmost layer as a nearest neighbor to the NC (now buried in the first sublayer). This NVH complex is even more stable than NC on the same layer. Third, H protects the already formed NV centers by forming low-energy NVHX complexes. These NV centers with their axes pointing along the directions of surface C–H bonds during their incorporation explain the experimental observations by CVD growth on (1 0 0) and (1 1 0) surfaces. Based on the model, we predict that CVD growth on (1 1 1) surface could eliminate the orientation domains to significantly improve the performance of NV centers.
Export citation and abstract BibTeX RIS
For more information on this article, see LabTalk.
1. Introduction
Because of the unique spin and optical properties, nitrogen-vacancy (NV) center in diamond is a promising candidate for many attractive applications such as quantum information processing, magnetometry, and electric sensing [1–4]. Due to its quantum mechanical nature, manipulating the location, population, and orientation of these NV centers can be crucial for the success of such applications [5, 6]. Thus, it is paramount important to achieve a precise control of the formation of the NV centers in diamond. To date, nitrogen ion implantation and chemical vapor deposition (CVD) are the two widely deployed experimental methods for NV centers. The CVD method is believed to be superior to the nitrogen ion implantation method, because the latter will also cause uncontrollable crystal damages that seriously degrade the performance of the NV centers [7–10].
At present, the formation mechanism of NV centers by ion implantation has been generally agreed upon. First, the implanted N atoms incorporate onto the substitutional sites (NC). With subsequent annealing (>600 °), the NC's capture nearby vacancies (VC), which are highly mobile, to form the NV centers [8–13]. On the other hand, the incorporation of the NV centers in the CVD method is not yet well understood. Two possible mechanisms have been proposed [12, 13]: one is characterized by the capture of vacancy, similar to that of ion implantation, while the other focuses on the effect of surfaces where the NV centers form as a natural result of epitaxial growth. A number of experiments support the notion that the formation of the NV centers is a surface-related phenomenon [12, 14, 15]. In particular, it was reported that NV centers grown on diamond (1 1 0) surface show preferential orientations along only two of the four possible crystallographic axes [12, 14]. Interestingly, a similar orientation preference was also observed for Si-related impurities grown in diamond [15]. In other words, only the interaction between diamond surfaces and impurities may explain the incorporation mechanism of NV centers in the CVD method. Besides, during the incorporation of the NV centers, other kinds of defects and impurities, such as NC, VC and nitrogen-vacancy-hydrogen (NVH) complexes, are also introduced [12–14, 16]. Unintentional defects/impurities could degrade the performance of the NV centers. Thus, understanding the fundamental physics of the impurities and maximizing the yield of NV centers during growth are crucially important for the applications of the NV center-based technologies.
In this paper, we propose a growth model for NV centers incorporated during CVD growth of diamond, based on first-principles calculations. A key factor of the model to nucleate and stabilize the NV centers is the presence of H. The model contains three steps: at first, an NC is formed favorably on the top surface layer, substituting a surface CH group. In the subsequent growth, a VC is preferably formed at the newly formed surface layer as the nearest neighbor to the NC (now at the first sublayer), forming an NV center along one of the dangling bond directions of the surface. Thus, on the (1 0 0) surface, there are four possible orientations for the NV centers, whereas on the (1 1 0) and (1 1 1) surfaces, the respective symmetries determine that there are only two and one possible orientations for the NV centers, respectively. These surface-born NV centers are subsequently buried into diamond lattice with further deposition. The model applies to NV centers grown in diamond on all surfaces. Not only the results agree with available experimental observations on the (1 1 0) and (1 0 0) surfaces, it further predicts that, on the (1 1 1) surface, there should be only NV centers along one orientation, which could greatly improve the sensitivity of the grown NV centers.
2. Model and method
Spin-polarized density-functional theory calculations are performed with the Vienna Ab-initio simulation package (VASP) [17]. The Kohn–Sham wave functions are expanded in a plane wave basis set with a cut-off energy of 400 eV. The projector-augmented wave (PAW) method and PBE potential for the exchange-correlation functional are used [18]. A diamond surface is simulated by a slab with 4 × 4 surface cell, 12C layers, and a vacuum layer ⩾12 Å. The bottom of the slab is passivated by H. The 2 × 2 × 1 M-P k-point mesh is used, which has been shown to yield good convergence [19, 21]. All the atoms in the slab except in the two bottom layers are allowed to fully relax until the forces acting on them are less than 0.02 eV Å−1.
An H-rich environment is typically adopted in the CVD growth [12, 20]. Thus, we consider H saturated surfaces in our calculations. The formation energy of an impurity or defect at the diamond surface is given by:
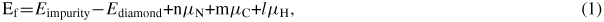
where Eimpurity is the total energy of the slab with an impurity (or defect), Ediamond is the total energy of the reference slab without the impurity, and n(m, l) is the number of N(C, H) atoms removed from the reference slab in order to incorporate the impurity. μN(μC, μH) is the atomic chemical potential of N(C, H). Under thermal equilibrium, μC should be the energy of C in bulk diamond. Since we consider the situation with maximum N concentration, μN can be taken as the energy of N in gas N2. Depending on the growth environment, μH may vary but should be within a specific range, e.g. from −0.37 eV determined by the formation of CH4 from diamond to 0 eV determined by the formation of H2.
3. Results and discussion
3.1. Growth model on (1 1 0) surface
The (1 1 0) surface is a popular substrate for growing NV centers by CVD [12–14]. Under H-rich growth condition, each C on the surface is passivated by one H and the degree of relaxations of surface C are highly reduced after passivation. The fact that the surface C atoms are close in their positions to their bulk counterparts indicates that the surface is fully stabilized by H passivation [21].
During CVD growth, impurities are formed more easily at surface than in bulk, with qualitatively different formation mechanisms [22, 23]. We consider three types of impurities, NC, VC and NVH complex, as the most important near-surface precursors for NV incorporation. On the H saturated surface layer, the most stable NC removes a C and a passivating H. The most stable VC has all three C dangling bonds passivated by H. This is different from bulk where the limited space prevents the H passivation of all four dangling bonds. A surface NVH, which is the most likely precursor to the NV centers, on the other hand, is composed of an NC, a VC and an extra H. The atomic structures and formation energies of surface impurities are shown in figure 1.
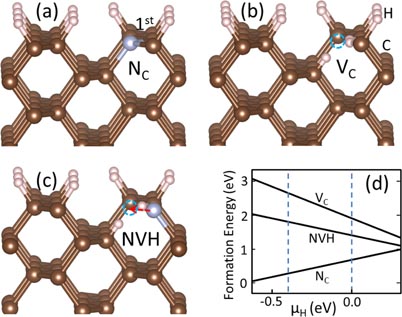
Figure 1. Atomic structures of (a) NC (light blue ball is N), (b) VC (dashed circle) and (c) an in-plane NVH complex at the top (1 1 0) surface layer, which is also the first layer of an epitaxial growth. Brown ball is carbon and smaller pink ball is hydrogen. Dashed arrow from NC to VC in the NVH complex denotes its orientation. (d) Impurity/Defect formation energy as a function of μH. Zero μH is for H in the state of H2, and −0.37 eV is for H in the state of CH4 with μC equals that of bulk diamond.
Download figure:
Standard image High-resolution imageIn the bulk, the NV centers are usually charged depending upon the position of the Fermi level of the system. In the initial growth at the surfaces, the most favorable structures of the N-related defects are usually neutralized by hydrogen atoms. Thus, charged NV complexes are not considered in this study.
The formation energies of surface impurities are indeed noticeably lower than those in bulk (
 ,
,
 and
and
 when μH = 0 eV). This is an indication that the incorporation of impurities during CVD growth should be more effective. Among the three impurities/defects, VC has the highest energy, NC has the lowest, while NVH is in the middle. If we assume that the growth mode of diamond is layer-by-layer, we can expect that at the first step of the NV center incorporation, an NC is formed favorably. Neither NVH nor VC will form in the first step. In other words, the growth of NV center depends on further deposition.
when μH = 0 eV). This is an indication that the incorporation of impurities during CVD growth should be more effective. Among the three impurities/defects, VC has the highest energy, NC has the lowest, while NVH is in the middle. If we assume that the growth mode of diamond is layer-by-layer, we can expect that at the first step of the NV center incorporation, an NC is formed favorably. Neither NVH nor VC will form in the first step. In other words, the growth of NV center depends on further deposition.
Figure 2 shows the possible defect structures when the next layer is grown on the already NC incorporated (1 1 0) surface. These structures either have low formation energies or are closely related to the formation of NV centers at the end. As shown in figure 2(a), the most stable structure is an NVH complex, composed of the NC (now in the first sublayer) and a VC in the newly grown surface layer. Typically, a surface layer containing no impurities (such as those in figures 2(b) and (c)) is not energetically favored. Hence, horizontal NV center in the first sublayer (namely, figure 2(b)) is also not favored. Increasing the number of passivating H could reduce the energy for the complex in figure 2(b) but it is still much higher than the one in figure 2(a), as shown in figure 2(d). This result suggests that the most stable NVH complex at this stage must be formed in two steps: first, in depositing the first layer, an N substitutes a surface C. Second, with the deposition of the second layer, a VC is formed in the newly formed surface layer as the nearest neighbor of the preceding NC.
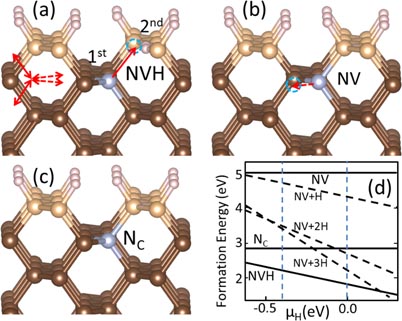
Figure 2. Atomic structures of the impurities when a second carbon layer (light brown balls) is deposited. (a) An out-of-plane NVH complex, (b) a buried in-plane NV center and (c) a buried NC, both in the first layer beneath the second deposited layer. Solid and dashed arrows (both single and double headed) stand for out-of-plane dangling bond directions and in-plane bond directions of sp3 diamond, respectively. (d) Defect formation energy as a function of μH. The dashed lines in (d) shows the formation energy change of the in-plane NV center with more passivating H atoms.
Download figure:
Standard image High-resolution imageIn figure 2(a), double arrows represent the four C–C bonds in diamond. On the (1 1 0) surface, two of them (the solid ones) give dangling bond directions out of the surface and the other two (dashed ones) are within the surface. Therefore, the discussion above asserts that possible orientations of the NVH complexes on the (1 1 0) surface are those along the solid arrows only. This result explains the observed preferential orientations of NV centers in [12]. It also indicates that near surface NVH complexes are responsible for the NV centers formed inside bulk (although the latter maybe only a fraction of the former).
To better understand the role of surface, figure 3 shows the local density of states (LDOS) of C and N in the vicinity of impurities in the bulk and at surface. We note that in the bulk, VC has deep gap states originated from C dangling bonds; NC is a relatively shallow donor with occupied anti-bonding states close to the conduction band minimum (CBM). These high-lying states are the reasons why isolated VC and NC have relatively high energies. Figure 3(e) shows that an NV center also consists of deep gap states but the N-related states are now near the valence band maximum (VBM). The net result of NV formation is thus to significantly lower the energy of occupied N states to lower system total energy. It explains why NC attracts VC in bulk diamond.

Figure 3. Local density of states (LDOS) of ((a) and (b)) VC, ((c) and (d)) NC and ((e) and (f)) NV center in bulk, and at (1 1 0) surface, respectively. The corresponding Fermi levels are indicated by vertical dashed lines. Shaded areas are the LDOS of bulk diamond, with the VBM and CBM indicated.
Download figure:
Standard image High-resolution imageThe situation is completely different at the surface. For the VC, figure 3(b) shows that the deep gap states have completely gone. This is because the dangling bonds of the VC have all been passivated by H. For NC, figure 3(d) shows that the originally near-CBM states are now below the VBM. This is because the NC has a dangling bond to accommodate the one extra electron. The resulting surface NVH also has no gap states, as revealed by figure 3(f). It is this surface effect that makes the formation of NC and VC, as well as that of NVH complex in CVD diamond considerably more favorable. Note that figure 1(d) shows that a fully passivated VC is still relatively high in energy than an NC. This could be a strain effect as VC has a limited space to host three H atoms.
Until now, the incorporation of NV centers is not completed yet. With more layers deposited, the atomic structure of the N-related impurities may still change, e.g. an incoming C could occupy a VC site in NVH, thereby reducing the complex back to the original NC. Figure 4 shows the representative atomic structures of the formed possible complexes, and the corresponding formation energies. We see that an NV center straddling the first and second sublayers without any H, has the highest energy. With passivating H (denoted as NVHX, where X = 1–3 is the number of H), the energy could be lowered. Generally, the higher the H chemical potential, the lower the formation energies of NVHX, among which NVH2 is the most stable. When the H chemical potential is low, on the other hand, reduced NC can have the lowest energy. These suggest that, in CVD growth, there is a delicate balance for the choice of μH to avoid having either too many hydrogenated NV centers or too many reduced NC. Note that, unlike at the top surface, here the dangling bonds of NVH2 are not fully passivated due to limited vacancy space. Experiments showed that NV centers in diamond are always accompanied by other defects, of which the most popular ones are NC and NVHX [12–14, 16] with a typical concentration ratio NC : NVHX : NV = 300 : 30 : 1. It qualitatively agrees with our calculated results at low μH in figure 4.

Figure 4. Atomic structures of the impurities when a third carbon layer (light gray balls) is deposited. (a) Out-of-plane NVH3 complex and (b) out-of-plane NV center formed following the growth of the NVH complex in figure 2(a). (c) Defect formation energy as a function of μH.
Download figure:
Standard image High-resolution imageThe properties of the NVHX complexes have been studied experimentally [12, 13, 16]. Usually, hydrogenated NV centers are expected to contain only one H (bonded to a C). Commonly used experimental techniques for studying NV centers usually involve electron paramagnetic resonance or optical spectroscopic techniques. However, these techniques are not so sensitive to local structures of the NVHX. Based on the calculated formation energy, over a wide range of general growth conditions, we find X = 2 is most stable, followed by X = 3 and then by X = 1. The energy differences (∼ 1 eV) are not small. Of course, at elevated temperature for diamond growth, one may also need to take into account the effect of entropy which would favor X = 1.
3.2. On (1 0 0) and (1 1 1) surfaces
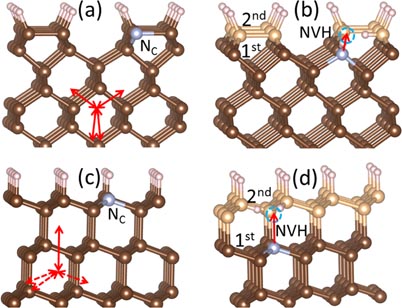
Besides the (1 1 0), one may also use (1 0 0) or (1 1 1) surfaces for CVD growth [12–14, 19, 20]. For (1 0 0) surface, the most stable structure contains 2 × 1 dimer rows, in which every surface C binds to one H [23]. For (1 1 1) surface, the most stable structure is 1 × 1 where every surface C binds to one H [19]. Similarity of these hydrogenated surfaces suggests that the above (1 1 0) surface-assisted multi-step growth model for NV centers may also apply to (1 0 0) and (1 1 1) surfaces. Indeed, figures 5(a) and (c) show that an NC can easily form at the topmost (1 0 0) and (1 1 1) surfaces similar to (1 1 0) surface. Figure 5(b) shows that, as the growth proceeds on the (1 0 0) surface, a vacancy can be easily formed adjacent to the NC now at the first sublayer. The (1 1 1) surface is, however, slightly different, as growth may proceed in a double-layer-by-double-layer fashion. Figure 5(d) shows that under the double-layer growth mode, once the NC is formed (now at the top part of the first sub bilayer), both NVHX and NV could form in subsequent growth.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 5. Atomic structures of (a) an NC and (b) an NVH complex on the (1 0 0) surface and (c) an NC and (d) an NVH complex on the (1 1 1) surface. Solid arrows indicate the possible orientations of the NV centers, while dashed arrows indicate the impossible ones. The growth sequence is denoted by first and second single (for (0 0 1)) and double (for (1 1 1)) layers.
Download figure:
Standard image High-resolution image{kind=link}
On the (1 0 0) surface, the dangling bonds have four directions, as shown by double arrows in figure 5(a). At any time, two equivalent directions appear on the top surface, but as the growth proceeds, the other two directions take over. This process repeats. Hence, over time, all four directions are equivalent, so one can expect that there are four equivalent NV axes. This result agrees with experiment [14]. On the other hand, on the (1 1 1) surface, due to the doubly-layer growth mode, the direction of the dangling bonds is only along one axis, namely, along [1 1 1] at all times.
Thus, to incorporate the NV centers during CVD growth, on the (1 1 0) and (1 0 0) surfaces, one needs three separate steps, involving three sequential atomic layers. On the (1 1 1) surface, one needs, on the other hand, only two steps, involving two sequential atomic double-layers. Although detailed growth kinetics could be different on different surfaces to result in different concentration ratios among NC, NV and NVH, the orientation dependence of the NV centers, which is analyzed here solely based on local energetic consideration with demonstrated success when compared with available experiments, should remain the same, irrespective of the kinetics.
NV centers with only one orientation have many advantages in terms of technological applications, such as the improvement of magnetic sensitivity [14]. Therefore, CVD growth of NV centers on (1 1 1) surface is highly desirable [24, 25]. The growth model developed here should also apply to other semiconductors, as a means to control the incorporation of desirable impurities. In this regard, we note that Si-related complexes in diamond also show surface-dependent properties [15].
4. Conclusion
In summary, first-principles study reveals the orientation dependent of NV centers in CVD grown diamond. In the multi-step growth model, the incorporation of the NV centers is enhanced by H. Despite the simplicity of the model, it explains the formation of NV centers with four possible orientations in (1 0 0)-grown diamond and two possible orientations in (1 1 0)-grown diamond. These results agree with available experiments, demonstrating the validity of the model. Given the similarities in the NV center formation and incorporation near surfaces, we propose that using the (1 1 1) surface, one should be able to grow NV centers with only one orientation, which could be a significant step forward toward fundamental study of the NV centers as well as for technological applications.
Acknowledgments
The work is supported by the NSF of China with grant nos. 11074128, 11274179 and 91227101, and National 973 projects of China with nos. 2011CB922102 and 2012CB921900. SBZ was supported by the Department of Energy under grant no. DE-SC0002623.