High-Pressure Synthesis and Chemical Bonding of Barium Trisilicide BaSi3
by
 , ,
, ,
Julia-Maria Hübner
 ,
,
Lev Akselrud
,
Walter Schnelle
,
Ulrich Burkhardt
,
Matej Bobnar
,
Yurii Prots
,
Yuri Grin
and
Ulrich Schwarz
* Max-Planck-Institut für Chemische Physik fester Stoffe, Nöthnitzer Straße 40, 01187 Dresden, Germany
*
Author to whom correspondence should be addressed.
Materials 2019, 12(1), 145; https://doi.org/10.3390/ma12010145
Submission received: 30 November 2018
/
Revised: 22 December 2018
/
Accepted: 26 December 2018
/
Published: 4 January 2019
(This article belongs to the Special Issue Advances in Zintl Phases)
Abstract
:BaSi3 is obtained at pressures between 12(2) and 15(2) GPa and temperatures from 800(80) and 1050(105) K applied for one to five hours before quenching. The new trisilicide crystallizes in the space group I2m (no. 121) and adopts a unique atomic arrangement which is a distorted variant of the CaGe3 type. At ambient pressure and 570(5) K, the compound decomposes in an exothermal reaction into (hP3)BaSi2 and two amorphous silicon-rich phases. Chemical bonding analysis reveals covalent bonding in the silicon partial structure and polar multicenter interactions between the silicon layers and the barium atoms. The temperature dependence of electrical resistivity and magnetic susceptibility measurements indicate metallic behavior.
1. Introduction
The Zintl-Klemm concept [1,2] constitutes a powerful framework for understanding the interdependence of chemical bonding and electron count of a rich variety of binary phases formed by element semiconductors such as silicon or germanium with electropositive partners of the alkaline-, alkaline earth- and rare-earth metal groups. Counting rules for compounds such as Ba2Si [3], Ba3Si4 [4] and BaSi2 [5] work successfully when a complete charge transfer from the electropositive metal to the tetrel atoms is assumed. The formation of covalent two-center two-electron interactions in the resulting polyanionic partial structures yields an electron-precise electron balance. Phases violating the classical electron count because of unusual coordination environments in the covalent partial structure of the p-block element exhibit more exotic bonding properties, often in combination with metal-type electrical conductivity [6,7]. Quite a few of these so-called covalent metals are accessible by high-pressure high-temperature synthesis.
A systematic study of tetrel connectivities in polyanions revealed a variety of motifs with composition MT3 (M = Ca, Sr, Ba, Y, La, Ce, Eu, Gd, Tb, Ho, Er, Tm, Yb, Lu; T = Si, Ge) [8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27]. All of these exceed the scope of the 8-N rule and show interesting physical properties such as superconductivity. Although barium and germanium form two superconducting BaGe3 modifications, hP8 [14] and tI32 [15], a corresponding barium–silicon phase remained clandestine so far. In this study, we describe high-pressure high-temperature synthesis, crystal structure and chemical bonding properties of BaSi3 as well as the temperature dependence of electrical resistivity and magnetic susceptibility.
2. Materials and Methods
Barium trisilicide was prepared by high-pressure high-temperature synthesis. Sample handling except for high-pressure synthesis itself was accomplished in argon-filled glove boxes (MBraun, H2O and O2 <0.1 ppm). The precursor mixture was manufactured by arc melting of barium (Alfa Aesar, 99.9%) and silicon (Chempur, 99.999%) in the ratio 1:3 plus 1% excess of barium. The resulting material was intensely ground and placed in boron nitride crucibles before being transferred into MgO octahedra with an edge length of 14 mm. High-pressure high-temperature synthesis in a multi-anvil Walker-type module for one to five hours was realized at pressures between 12(2) and 15(2) GPa and temperatures from 800(80) and 1050(105) K before quenching to ambient temperature under load [28]. Calibrations of pressure and temperature by observing resistance changes of bismuth [29], as well as thermocouple-calibrated runs, had been conducted before the synthesis experiments.
Differential scanning calorimetry (DSC) experiments were realized in a Netzsch DSC 404 C device (Netzsch-Gerätebau GmbH, Selb, Germany) operated with heating and cooling rates of 10 K/min under argon atmosphere using corundum crucibles.
Phase designation was conducted by X-ray powder diffraction experiments with a Huber Image Plate Guinier Camera G670 (Huber Diffraktionstechnik GmbH & Co. KG, Rimsting, Germany), using CuKα1 radiation, λ = 1.54056 Å. High-resolution X-ray diffraction experiments of BaSi3 were conducted with synchrotron radiation (λ = 0.399972(2) Å) at beamline ID22 of the European Synchrotron Radiation Facility. All crystallographic calculations including the determination of diffraction peak positions as well as lattice parameter and crystal structure solution and refinement by the Rietveld technique (Table 1 and Table 2) were performed with the WinCSD program package, version 2018 [30].
For metallographic analysis, samples were prepared by polishing with diamond powder disks (grain size 6, 3 and 0.25 μm) in paraffin. The investigation was performed with a Philips XL 30 scanning electron microscope (LaB6 cathode), comprising an EDAX Si(Li) detector for energy-dispersive X-ray spectroscopy (EDXS).
Electronic structure calculations and chemical bonding analysis were carried out with the experimentally determined lattice parameters and the refined atomic coordinates of an idealized crystal structure model without disorder. First, band structure calculations were performed with the TB-LMTO-ASA (TB: tight-binding, LMTO: linear muffin tin orbitals, ASA: atomic sphere approximation) program package [31]. In these computations, the Barth-Hedin exchange potential [32] was used. The following radii of the atomic spheres were applied for the calculations: r(Ba1) = 2.375 Å, r(Ba2) = 2.419 Å, r(Si1) = 1.430 Å, r(Si2) = 1.415 Å, r(Si3) = 1.418 Å. Due to the calculations already including corrections for the neglect of interstitial regions and partial waves of higher order [33], insertion of additional so-called empty spheres was not necessary. A basis set of Ba(6s,5d) and Si(3s,3p) orbitals was employed for self-consistent calculations with Ba(6p,5d) and Si(3d) functions being downfolded. To obtain the partial waves, the radial scalar-relativistic Dirac equation was solved. After convergence, the electronic density of states (DOS) was calculated using a mesh of 32 × 32 × 32 k-points.
For the analysis of the chemical bonding in direct space the electron density and the electron localizability indicator ELI-D was calculated [34,35] with a module implemented in the program package. The computed spatial arrangement of ELI-D and electron density was analyzed with the program DGrid [36]. For this purpose, the electron density was integrated within so-called basins, i.e., space regions confined by zero-flux surfaces of the gradient field. This technique follows the procedure proposed in the Quantum Theory of Atoms In Molecules (QTAIM [37]) and provides electron counts for the basins of atoms (QTAIM populations of the atoms) and bonds (bond populations). The combined analysis of electron density and ELI-D constitutes a basis for the description of chemical bonding [38,39], especially in intermetallic compounds [40,41].
Electrical resistivity ρ was measured using a cuboid (1.00 mm × 1.80 mm × 0.90 mm) cut from a polycrystalline sample of cylindrical shape by a direct-current four-probe method carried out on a PPMS (Quantum Design International, San Diego, USA) AC transport option, 0.11 to 2.0 K and 2.0 to 305 K). The inaccuracy of ρ was estimated to be 20%, because of the intricate contact geometry. The measurement of magnetic susceptibility χ was conducted using a polycrystalline sample of cylindrical shape (diameter 1.0 mm, length 1.0 mm) and a SQUID magnetometer (MPMS XL-7, Quantum Design).
The thermal stability of the high-pressure phase was studied by differential scanning calorimetry (DSC) experiments. A commercially available Netzsch DSC 404C apparatus was equipped with corundum crucibles and operated with an argon atmosphere. Both heating and cooling were realized at a rate of 10 K/min.
3. Results and Discussion
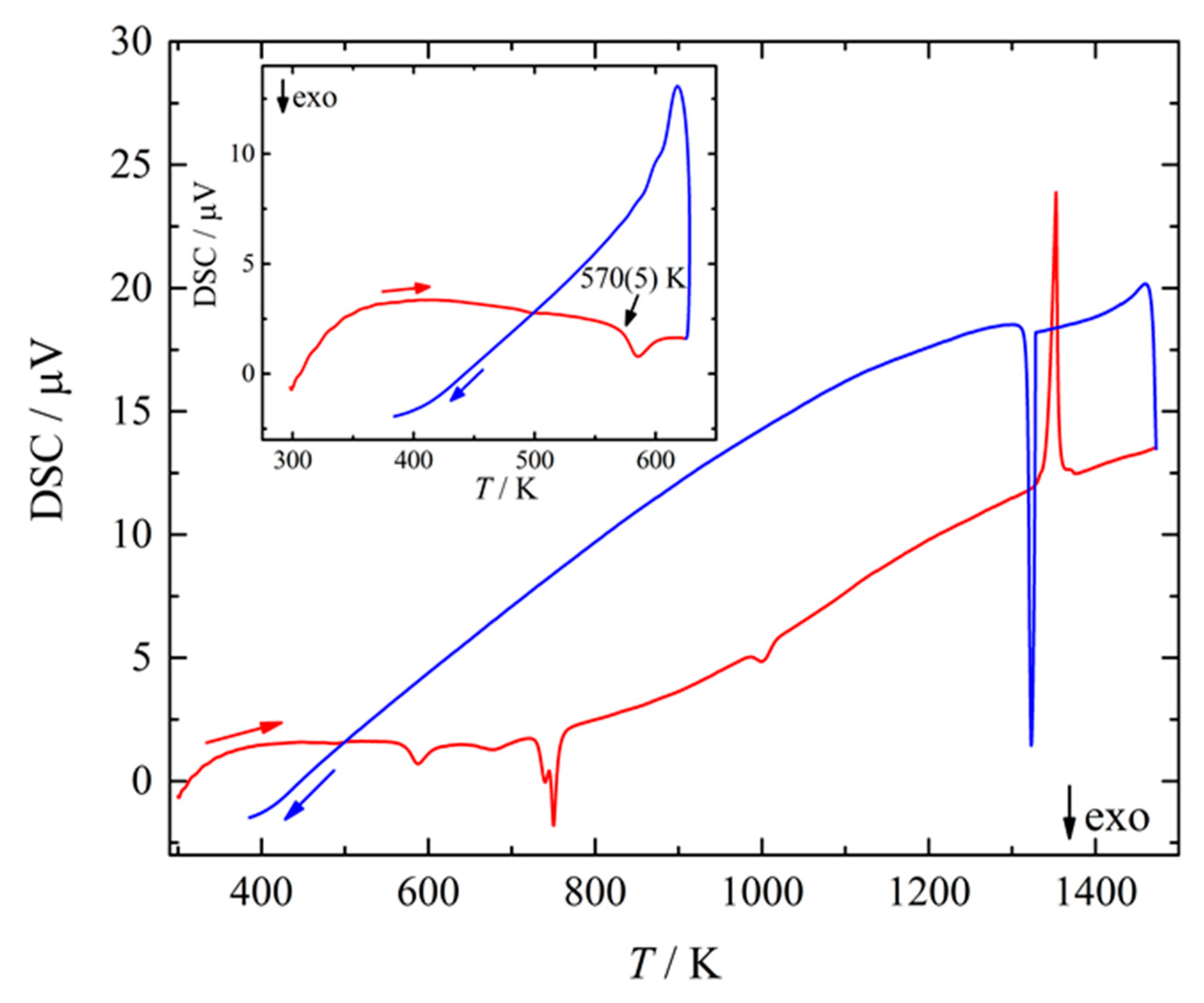
The new phase was obtained by high-pressure high-temperature treatment of pre-reacted Ba25Si75 mixtures before quenching. The chemical composition of the hp-ht product as determined using energy dispersive X-ray spectroscopy amounted to Ba22.4Si77.6 which corresponds to BaSi3 within the estimated error. Differential scanning calorimetry (DSC) measurements at ambient pressure evidenced the decomposition of BaSi3 (Figure 1) at 570(5) K. The first exothermic anomaly upon heating (Figure 1, inset) corresponded to the onset of disintegration. Powder XRD and EDXS analyses of the obtained decomposition product after heating to 623 K evidenced the formation of (hP3)BaSi2 [42] plus two amorphous phases with averaged composition Ba24.3(5)Si75.7(5) (≈BaSi3) and Ba14.5(5)Si85.5(5) (≈BaSi6), respectively. The following features at 725(5), 745(10) and 985(10) K represented different reaction steps of the decomposition products of BaSi3, and XRD data evidenced the formation of (oP24)BaSi2 (often labeled as Ba2Si4 [43]) and (cF8)Si [44]. As the final effect at 1325 K correlated with a corresponding signal upon cooling, the signal is essentially assigned to the eutectic of BaSi2 + Si in full accordance with phase diagram data [45]. As the cooling curve shows no further signal indicating the back transformation of BaSi2 and Si into BaSi3, the experimental data indicate that BaSi3 is a high-pressure phase, which is metastable at ambient pressure.
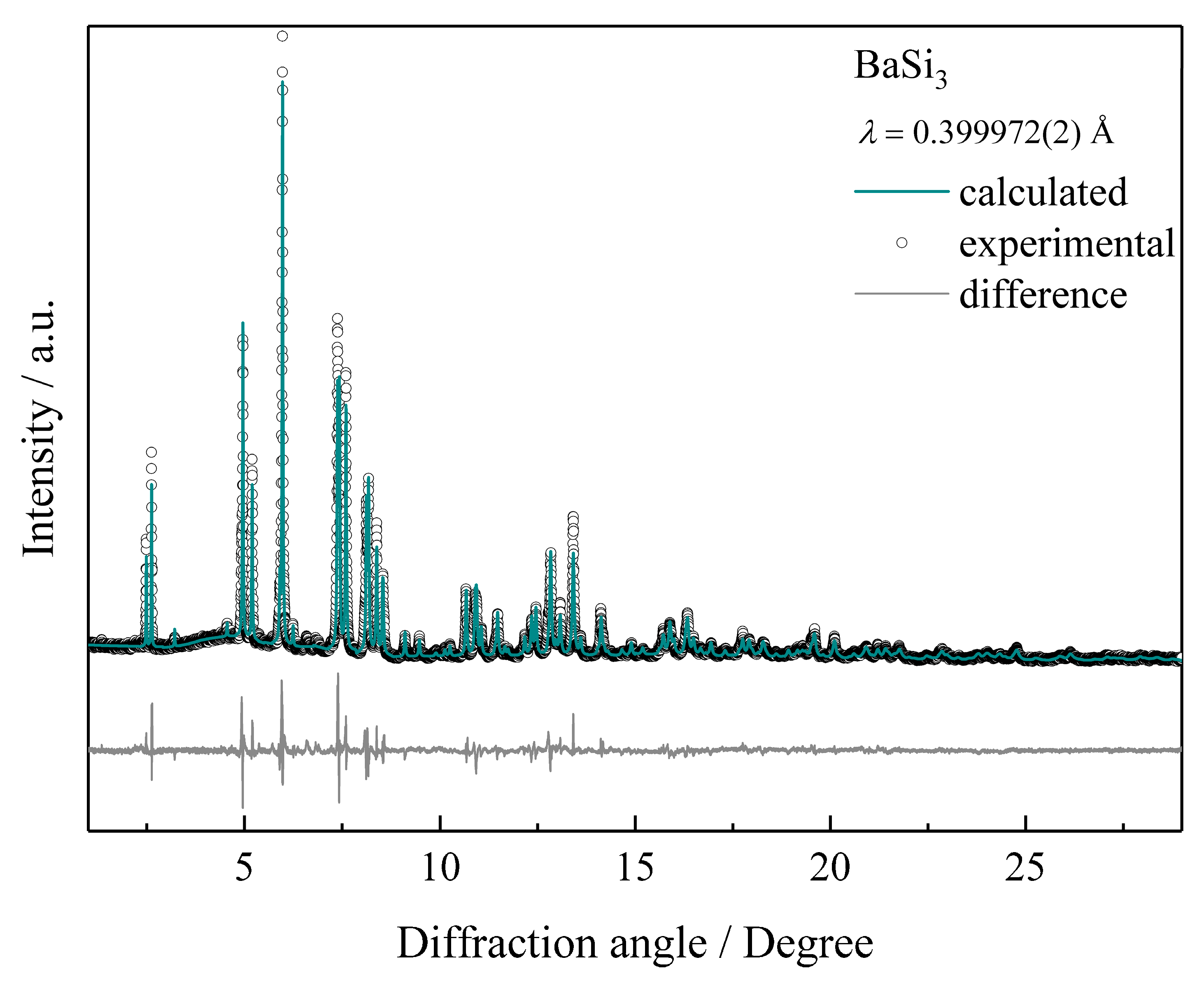
Characterization of the crystal structure was performed by X-ray powder diffraction experiments using synchrotron radiation. Indexing of peak positions yields a tetragonal unit cell for the new high-pressure phase BaSi3. The resulting diffraction symbol, as well as the axial ratio c/a, could be compatible with a CaGe3-type atomic arrangement as predicted by an earlier ab-initio study [46]. However, refinements of this structure pattern in space group I4/mmm (no. 139) do not produce satisfactory results with respect to reflection intensities (which is reflected in residuals R(P) = 0.215). Thus, structure models in maximal non-isomorphic subgroups were developed. The refinement of a structure model in space group I2m converged in a straightforward manner (R(P) = 0.073). However, unusually large displacement parameters motivated the introduction of split-positions for Ba1 and Si3 in the final refinements (Figure 2 and Table 1 and Table 2). As the profiles of some reflections show evidence for subtle shoulders, crystal structure solutions assuming further decrease of symmetry were attempted. With the available X-ray powder diffraction data, these tests remained unsuccessful.
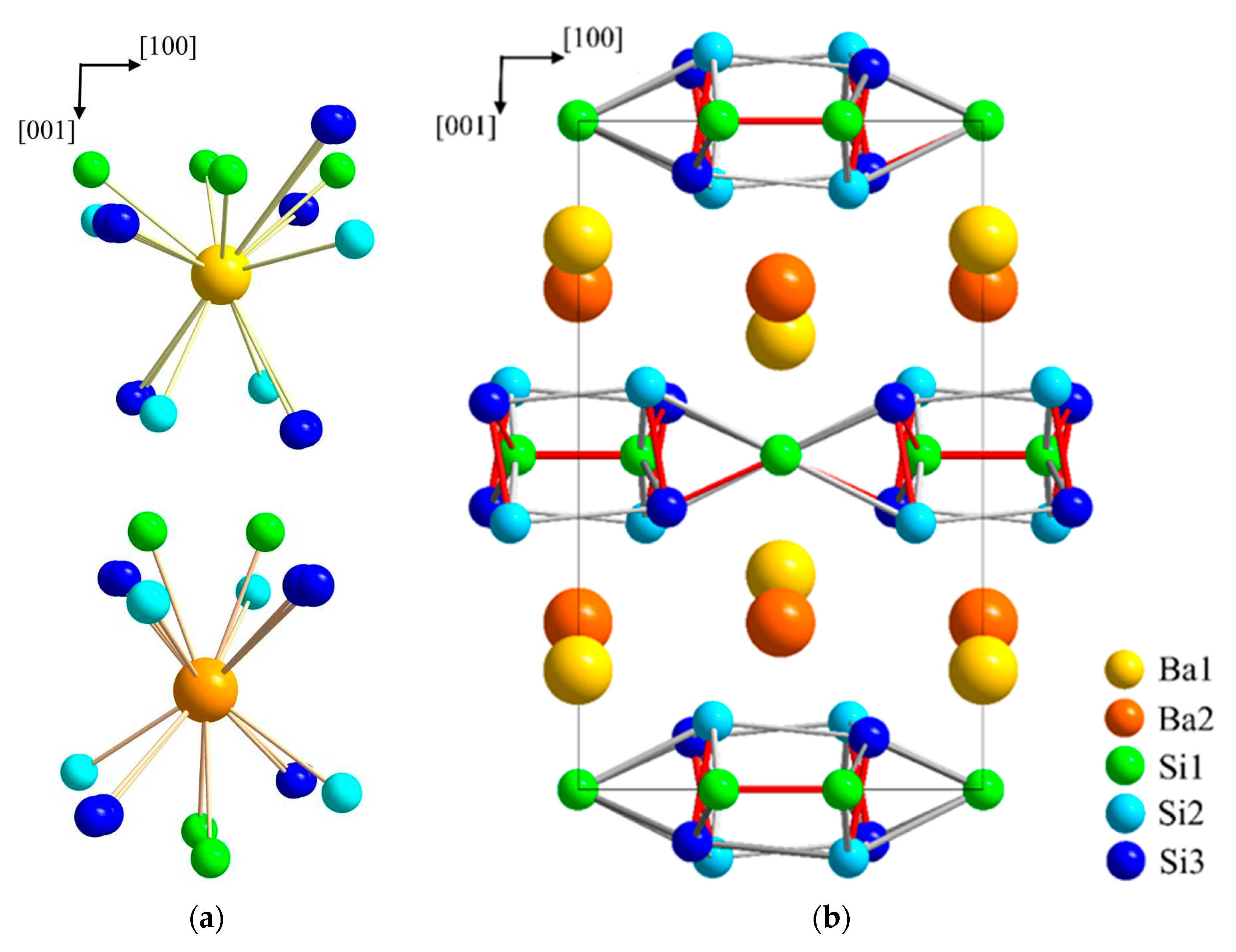
The crystal structure of BaSi3 (Figure 3) may be described as comprising silicon layers which are stacked along the c axis and separated by barium atoms. Ba1 and Ba2 are surrounded by 13 and 12 silicon atoms, respectively. The barium–silicon distances in the irregular polyhedrons of Ba1 and Ba2 cover the range from 3.325(8) to 3.79(1) Å and from 3.375(3) to 3.592(9) Å, respectively. For comparison, the binary silicon-rich barium compounds BaSi6 and BaSi2 exhibit Ba-Si distances in the range from 3.20(1) to 3.82 Å [47,48,49,50].
The silicon atoms occupy three distinct positions. The shortest distances d(Si1-Si1) of 2.34(1) Å and d(Si2-Si3) of 2.33(2) Å denote the occurrence of Si2 entities which are similar to the dumbbells occurring in CaGe3. However, in BaSi3 these primary Si2–Si3-fragments are inclined by approximately 10° with respect to the c axis. The tilt causes a 5° twist of the rectangular units in b direction with alternating rotation directions of neighboring units. The resulting distorted tetragonal prisms are linked by (Si1)2-dumbbells in a perpendicular orientation. As a result of the tilting, the distances d(Si1-Si3), which are of equal length in the CaGe3 type, distort into short (2.351(8) Å) and longer contacts (2.524(8) Å).
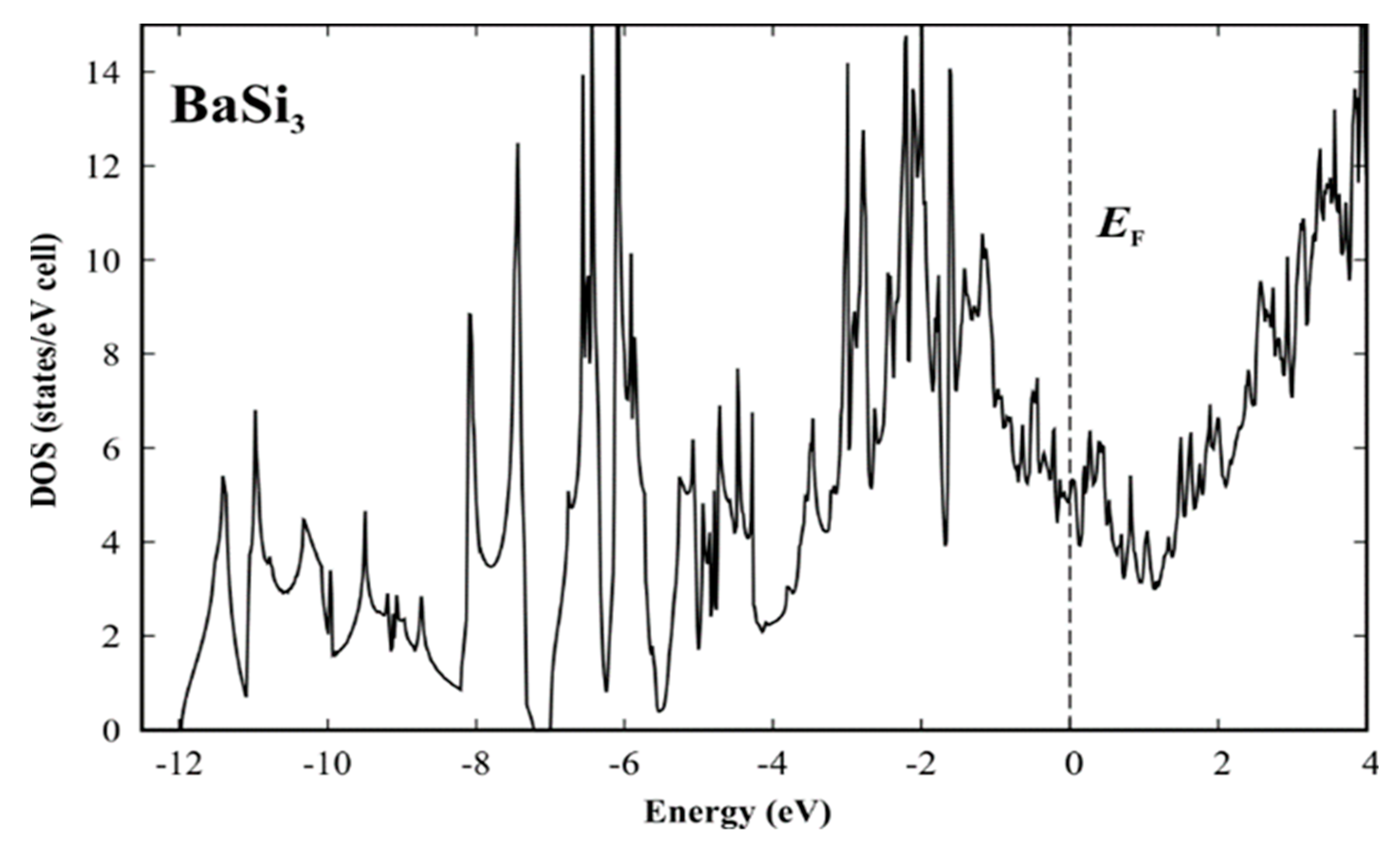
The refined crystal structure model of BaSi3 evidences that the new compound comprises silicon atoms in unusual connectivity situations. Assuming single-bonded silicon dumbbells would imply a rather non-realistic electron balance with huge electron demand: (Ba2+)2{[(1b)Si−(1b)Si)]6−}3 × 12p1+. Considering the slightly longer distances of Si1 and Si3 as additional single bonds reduces the problem: (Ba2+)2[(3b)Si11−]2[(1b)Si23−]2[(3b)Si31−]2 × 6p1+. In agreement with this predicted electron demand, the calculated electronic density of states reveals that the Fermi level (calculated for the idealized structure model without disorder [51]) is located below the pseudogap (Figure 4). Nevertheless, a quantitative estimate of the electron count requires a more elaborate analysis of chemical bonding in real space [34,35].
By comparison with distances in analogous compounds (d(Si-Si) between 2.390(1) and 2.443(3) Å [8,9,10]) or modifications of elemental silicon (d(Si-Si) in cI16 from 2.3283(4) to 2.3841(4) Å [52] and in cF8 2.3516 Å [53]), the shortest homoatomic Si-Si contacts in BaSi3 (2.33(2) to 2.351(8) Å) may easily be considered as bonding. The remaining interactions in BaSi3 require further analysis.
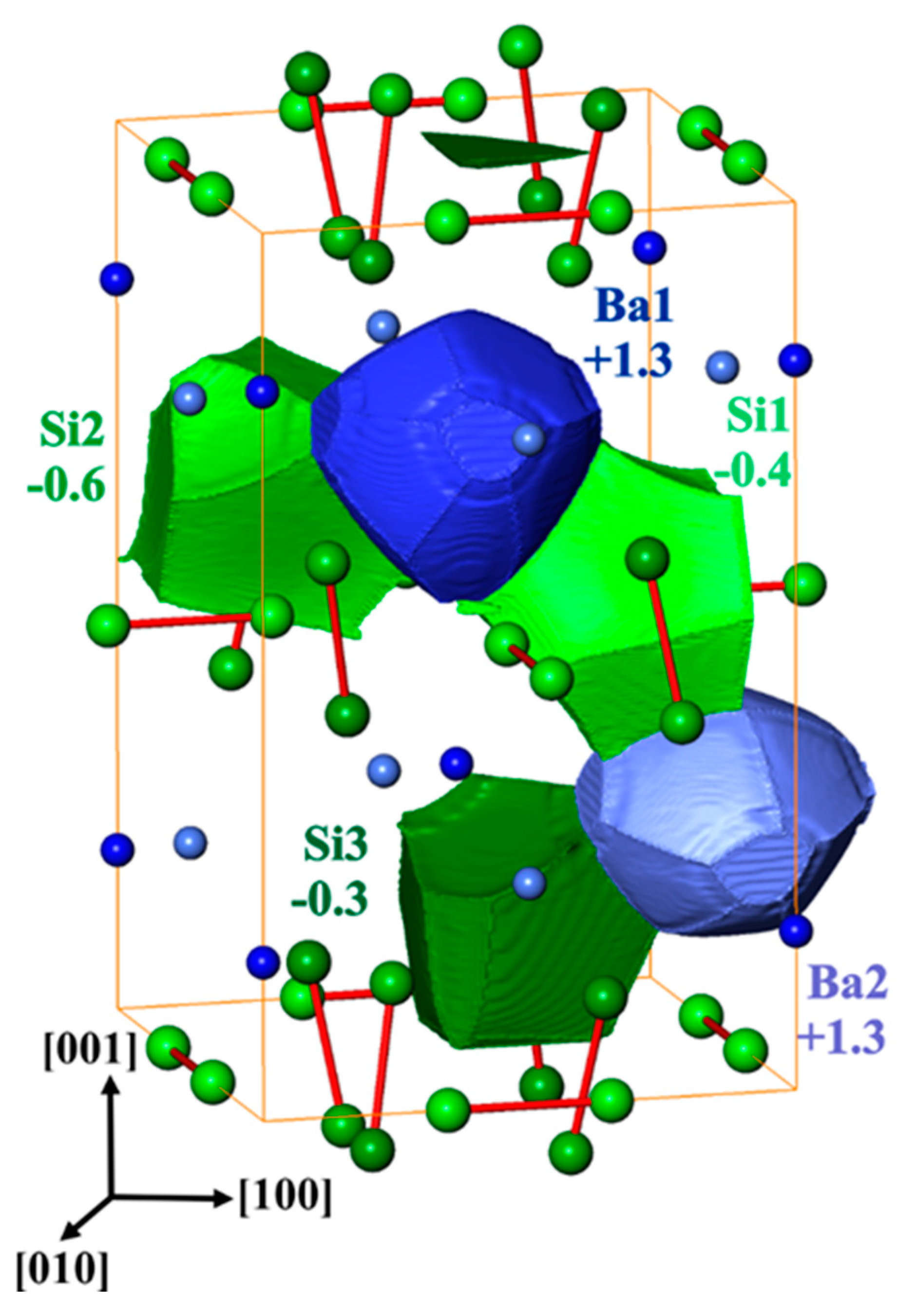
The calculated electron density reveals Ba species with almost spherical shape indicating essentially ionic character. The shapes of the silicon species have a more polyhedral character, especially the contact faces between the two nearest silicon atoms appear rather flat, which is characteristic for non-polar covalent bonding. Integrating the electron density within the QTAIM shapes and subtracting the atomic number yields effective charges. The net charge transfer from barium to silicon (Figure 5) is in accordance with the electronegativity difference of the constituents. The effective charges of the barium species (+1.30) fall into the range of +1.2 to 1.4 which is observed for barium–germanium clathrates [54] but are markedly smaller than those of calcium in CaSi3 (+1.44, +1.49 [8]). Moreover, the computed charges of silicon in BaSi3 (−0.3 to −0.6) show a larger spread than those in CaSi3 (−0.40 and −0.54). Both findings consistently indicate a slightly different organization of the bonding in BaSi3 in comparison to the other trisilicides.
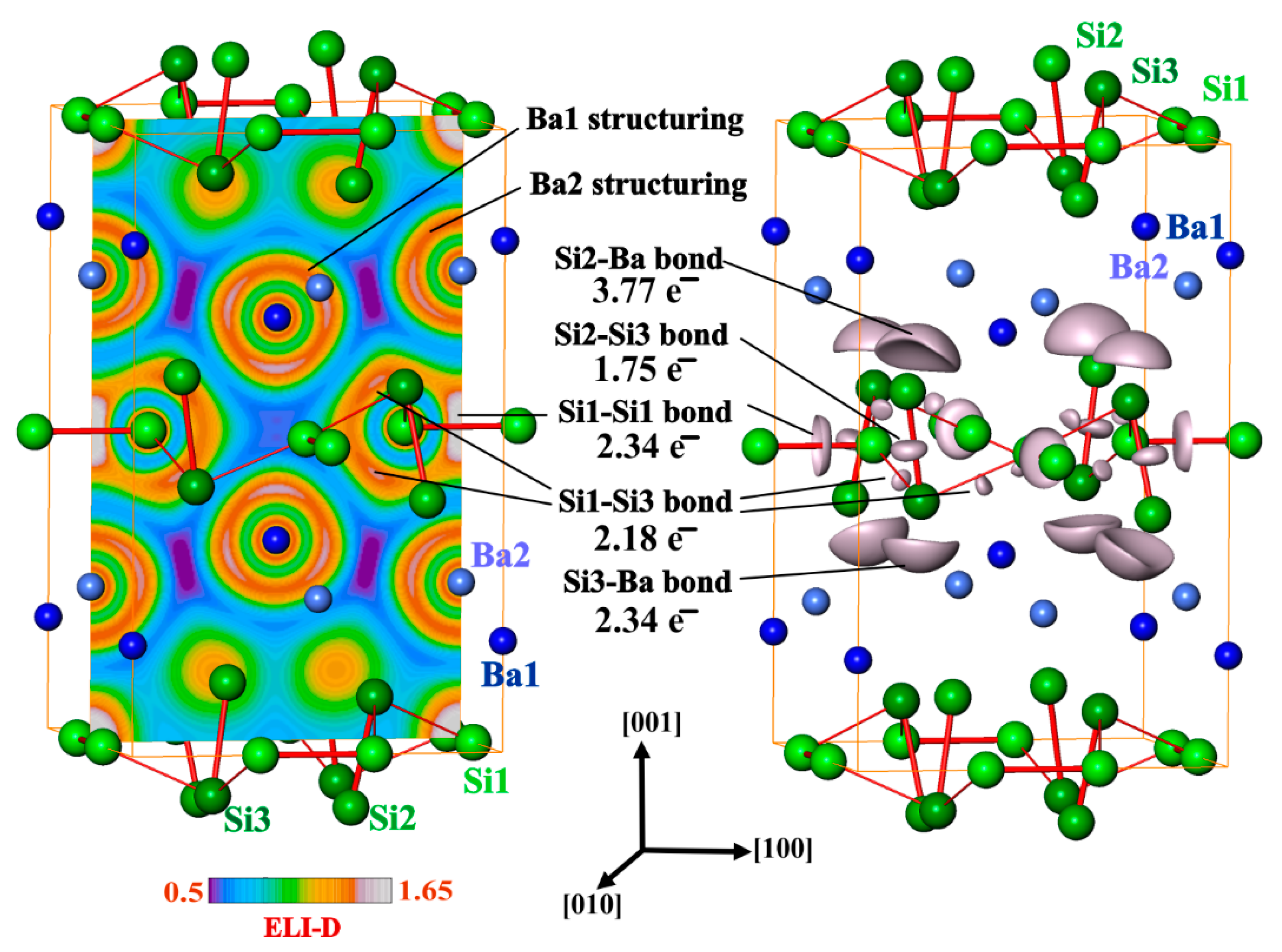
Further analysis of the chemical bonding in BaSi3 was realized by applying the electron localizability approach. The ELI-D distribution in the penultimate shell of the barium atoms shows significant deviations from spherical symmetry (structuring [38], Figure 6, left pannel). Quantitative characterization of this trend and comparison to the recent results for YGa6, YGa and t-Y5Ga3 [55] reveals fingerprints for the participation of the penultimate shell in the bonding interactions [38,56]. In the valence region of silicon, five different ELI-D maxima are observed. Three of them are located on (or close to) the shortest Si-Si contacts (Figure 6) indicating covalent Si-Si bonding. Two remaining ones are located on the outer side of the Si2-Si3 dumbbell suggesting lone pairs. In an isolated Si2 molecule, each basins of these maxima would have contacts with one core basin of silicon, i.e., it would reflect a lone pair such as in RhBi4 [57], CoBi3 [58] or hp-CuBi [59]). In BaSi3, each of these attractors represents a five-center bond SiBa4.
Integration of the electron density within the bonding basins (Figure 6, right pannel) yields the values 2.34 and 1.75 for the Si1-Si1 and Si2-Si3 dumbbells, respectively, as well as 2.18 for the Si1-Si3 contact. Consequently, the shortest Si-Si distances correspond 2c-2e bonds in good approximation. The calculated population of the lone-pair basin of Si3 amounts to 2.34 electrons, which is close to the value of two as predicted by the 8-N rule. Yet, the three-bonded Si1 atom does not show any basin resembling a lone-pair. The Si2 atom is single-bonded with a calculated population of the lone-pair basin of 3.77 electrons, which is still below the predicted six on basis of the 8-N rule. The analysis evidences that the interpretation of the crystal structure of BaSi3 as a CaGe3-type packing with slightly tilted Si2 dumbbells is insufficient.
Instead, the bonding analysis yields direct evidence for a more adequate description of the atomic arrangement (Figure 7). The Si2 dumbbells (Si1)2 and Si2-Si3 are oriented in an almost perpendicular way. However, the condensation of these diatomic units proceeds exclusively via three-bonded Si1 and Si3 atoms. The Si2 atoms form a single bond to the Si3 atoms and are arranged above and below the puckered sheets formed Si1 and Si3. These special covalent segments are interconnected by barium cations interacting with the anionic substructure by polar five-center Si2Ba4 and Si3Ba4 bonds.
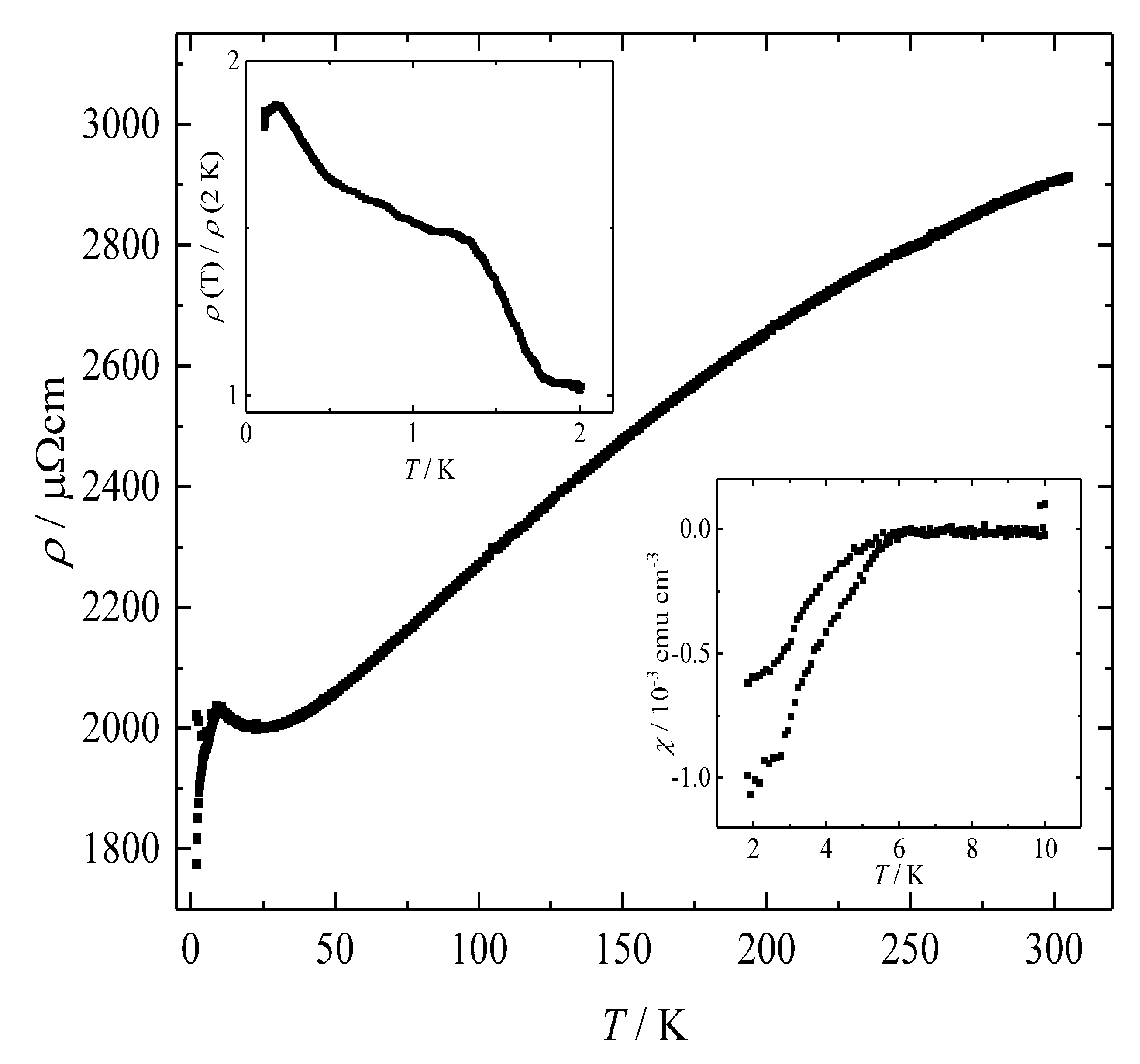
In agreement with the electron balance and the calculated band structure, electrical resistivity measurements on BaSi3 between 2 and 305 K show a positive slope above approximately 25 K indicating metal-type conductivity behavior (Figure 8). At room temperature and zero-field, the value for BaSi3 amounts to ρ300 K = 2906 μΩ cm with a residual resistance ratio (RRR) ρ293 K/ρ4 K = 1.5. In comparison to analogue high-pressure phases of germanium [12,13,19,27], the resistivity is high and shows only weak temperature dependence which is typical for polycrystalline silicides [60]. The changes of both resistivity and susceptibility below 6 K are attributed to traces of superconducting BaSi2 [61]. The little overall resistivity changes down to 110 mK in conjunction with the minor magnetic susceptibility change (Figure 8) clearly evidence that these changes do not originate from a bulk superconducting state.
Author Contributions
The concept of the study was developed by U.S. and J.-M.H.; the quantum chemical analysis was performed by Y.G.; X-ray diffraction experiments by Y.P.; structure refinements by L.A. and J.-M.H.; magnetic susceptibility and interpretation by M.B.; electrical resistivity and interpretation by W.S.; metallographic characterization by U.B.; writing—original draft preparation, J.-M.H., Y.G. and U.S.; writing—review and editing, U.S.
Funding
Financial support for J.-M.H. by the International Max Planck Research School for Chemistry and Physics of Quantum Materials (IMPRS-CPQM) is gratefully recognized. We acknowledge the European Synchrotron Radiation Facility for provision of synchrotron radiation facility at ID22.
Acknowledgments
We express our thanks to Susann Leipe and Liudmil a Muzica for supporting high-pressure syntheses. We like to thank Marcus Schmidt and Susann Scharsach for DSC measurements as well as Sylvia Kostmann and Petra Scheppan for metallographic characterizations. The assistance of Wilson Mogodi with synchrotron X-ray diffraction measurements at beamline ID22 of the ESRF is appreciatively recognized.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zintl, E.; Brauer, G.Z. Über die Valenzelektronenregel und die Atomradien unedler Metalle in Legierungen. Phys. Chem. B 1933, 20, 245–271. [Google Scholar] [CrossRef]
- Zintl, E. Intermetallische Verbindungen. Angew. Chem. 1939, 52, 1–6. [Google Scholar] [CrossRef]
- Widera, A.; Schäfer, H. Darstellung und Kristallstruktur des Ba2Si. Z. Naturforsch. 1976, 31, 1434–1435. [Google Scholar] [CrossRef]
- Eisenmann, B.; Janzon, K.H.; Schäfer, H.; Weiss, A. Zur Kenntnis von Ba3Si4. Z. Naturforsch. 1969, 24, 457–458. [Google Scholar] [CrossRef]
- Gladyshevskii, E.I. The crystal structure of BaSi2 and CeGe2. Dopovidi Akademii Nauk Ukrains’koi RSR 1959, 1959, 294–297. [Google Scholar]
- San-Miguel, A.; Toulemonde, P. High-pressure properties of group IV clathrates. Adv. High Pressure Res. 2005, 25, 159–185. [Google Scholar] [CrossRef]
- Flores-Livas, J.A.; Debord, R.; Botti, S.; San-Miguel, A.; Pailhès, S.; Marques, M.A.L. Superconductivity in layered binary silicides: A density functional theory study. Phys. Rev. B 2011, 84, 184503. [Google Scholar] [CrossRef]
- Schwarz, U.; Wosylus, A.; Rosner, H.; Schnelle, W.; Ormeci, A.; Meier, K.; Baranov, A.; Nicklas, M.; Leipe, S.; Müller, C.J.; et al. Dumbbells of Five-Connected Silicon Atoms and Superconductivity in the Binary Silicides MSi3 (M = Ca, Y, Lu). J. Am. Chem. Soc. 2012, 134, 13558–13561. [Google Scholar] [CrossRef]
- Meier, K.; Cardoso-Gil, R.; Schwarz, U. Crystal structure of holmium trisilicide, HoSi3. Z. Kristallogr. 2011, 226, 297–298. [Google Scholar] [CrossRef]
- Wosylus, A.; Prots, Y.; Schwarz, U. Crystal structure of ytterbium trisilicide, YbSi3. Z. Kristallogr. NCS 2011, 226, 295–296. [Google Scholar] [CrossRef]
- Schnelle, W.; Ormeci, A.; Wosylus, A.; Meier, K.; Grin, Y.; Schwarz, U. Dumbbells of Five-Connected Ge Atoms and Superconductivity in CaGe3. Inorg. Chem. 2012, 51, 5509–5511. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, T.; Fukuoka, H.; Inumaru, K. High-Pressure Synthesis and Electronic Structure of a New Superconducting Strontium Germanide (SrGe3) Containing Ge2 Dumbbells. Inorg. Chem. 2015, 54, 7433–7437. [Google Scholar] [CrossRef] [PubMed]
- Castillo, R.; Schnelle, R.; Baranov, A.I.; Burkhardt, U.; Bobnar, M.; Cardoso-Gil, R.; Schwarz, U.; Grin, Y. Trigermanides AEGe3 (AE = Ca, Sr, Ba): Chemical bonding and superconductivity. Z. Naturforsch. B 2016, 71, 585–592. [Google Scholar] [CrossRef]
- Castillo, R.; Baranov, A.; Burkhardt, U.; Cardoso-Gil, R.; Schnelle, W.; Bobnar, M.; Schwarz, U. Germanium Dumbbells in a New Superconducting Modification of BaGe3. Inorg. Chem. 2016, 55, 4498–4503. [Google Scholar] [CrossRef]
- Fukuoka, H.; Tomomitsu, Y.; Inumaru, K. High-Pressure Synthesis and Superconductivity of a New Binary Barium Germanide BaGe3. Inorg. Chem. 2011, 50, 6372–6377. [Google Scholar] [CrossRef]
- Belyavina, N.M.; Markiv, V.Y.; Speka, M.V. Crystal structure of YGe3, YGe1.9 and a novel Y3Ge4 compound. J. Alloys Compd. 1999, 283, 162–168. [Google Scholar] [CrossRef]
- Fukuoka, H.; Suekuni, K.; Onimaru, T.; Inumaru, K. High-Pressure Synthesis and Superconductivity of a New Binary Lanthanum Germanide LaGe3 with Triangular Ge3 Cluster Units. Inorg. Chem. 2011, 50, 3901–3906. [Google Scholar] [CrossRef]
- Fukuoka, H.; Yamanaka, S. High-Pressure Synthesis and Properties of a Cerium Germanide CeGe3 with the Cubic Cu3Au Type Structure. Chem. Lett. 2004, 33, 1334–1335. [Google Scholar] [CrossRef]
- Castillo, R.; Baranov, A.; Burkhardt, U.; Grin, Y.; Schwarz, U. Triangular Ge3 Units in a New Modification of EuGe3. Z. Anorg. Allg. Chem. 2015, 641, 355–361. [Google Scholar] [CrossRef]
- Savysyuk, I.A.; Gladyshevskii, E.I.; Gladyshevskii, R.E. Crystal Structures of RGe3 and RGe2 Compounds (R = Y, Gd, Tb). In Proceedings of the 7th International Conference on Crystal Chemistry Intermetallic Compounds, Lviv, Ukraine, 25–28 September 1999; p. PB17. [Google Scholar]
- Schobinger-Papamantellos, P.; Rodriguez Carvajal, J.; Buschow, K.H.J. The multiple q-vector incommensurate magnetic structure of TbGe3. J. Phys. Condens. Matter 2007, 19, 236201. [Google Scholar] [CrossRef]
- Schobinger-Papamantellos, P.; de Mooij, D.B.; Buschow, K.H.J. Crystal Structures of the Compound DyGe3. J. Alloys Compd. 1992, 183, 181–186. [Google Scholar] [CrossRef]
- Schobinger-Papamantellos, P.; Rodriguez Carvajal, J.; Tung, L.D.; Ritter, C.; Buschow, K.H.J. Competing multiple-q magnetic structures in HoGe3: I. The magnetic phase diagram of HoGe3. J. Phys. Condens. Matter 2008, 20, 195201. [Google Scholar] [CrossRef]
- Eremenko, V.N.; Obushenko, I.M. Phase Diagram of the Erbium-Germanium System. Sov. Non-Ferrous Met. Res. 1981, 9, 216–220. [Google Scholar]
- Fukuoka, H.; Yoshikawa, M.; Baba, K.; Yamanaka, S. Preparation and Structures of Lanthanoid Germanides, PrGe3.36, NdGe3.25, and TmGe3 with Double Square Ge-Mesh Structures. Bull. Chem. Soc. Jpn. 2010, 83, 323–327. [Google Scholar] [CrossRef]
- Harada, M.; Fukuoka, H.; Matsumura, D.; Inumaru, K. Structure and Chemical Bonding of Binary Ytterbium Germanides, Yb3Ge5 and YbGe3, Prepared by High-Pressure and High-Temperature Reactions. J. Phys. Chem. C 2012, 116, 2153–2158. [Google Scholar] [CrossRef]
- Hübner, J.-M.; Bobnar, M.; Akselrud, L.; Prots, Y.; Grin, Y.; Schwarz, U. Lutetium Trigermanide LuGe3: High-Pressure Synthesis, Superconductivity, and Chemical Bonding. Inorg. Chem. 2018, 57, 10295–10302. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.; Carpenter, M.A.; Hitch, C.M. Some simplifications to multianvil devices for high pressure experiments. Am. Mineral. 1990, 75, 1020–1028. [Google Scholar]
- Young, D.A. Phase Diagrams of the Elements; UC Press: Berkeley, CA, USA, 1991. [Google Scholar]
- Akselrud, L.; Grin, Y. WinCSD: software package for crystallographic calculations (Version 4). J. Appl. Crystallogr. 2014, 47, 803–805. [Google Scholar] [CrossRef]
- Jepsen, O.; Burkhardt, A.; Andersen, O.K. The Program TB-LMTO-ASA. Version 4.7; Max-Planck-Institut für Festkörperforschung: Stuttgart, Germany, 1999. [Google Scholar]
- Von Barth, U.; Hedin, L. A local exchange-correlation potential for the spin polarized case: I. J. Phys. 1972, C5, 1629–1642. [Google Scholar] [CrossRef]
- Andersen, O.K. Linear methods in band theory. Phys. Rev. 1975, B12, 3060–3083. [Google Scholar] [CrossRef]
- Kohout, M. A measure of electron localizability. Int. J. Quantum Chem. 2004, 97, 651–658. [Google Scholar] [CrossRef]
- Kohout, M. Bonding indicators from electron pair density functionals. Faraday Discuss. 2007, 135, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Kohout, M. DGrid, Versions 4.6-5.0; Radebeul, Germany, 2018. [Google Scholar]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1999. [Google Scholar]
- Wagner, F.R.; Bezugly, V.; Kohout, M.; Grin, Y. Charge decomposition analysis of the Electron Localizability Indicator: A bridge between the orbital and direct representation of the chemical bond. Chem. Eur. J. 2007, 13, 5724–5741. [Google Scholar] [CrossRef] [PubMed]
- Kohout, M.; Savin, A. Atomic shell structure and electron numbers. Int. J. Quantum Chem. 1996, 60, 875–882. [Google Scholar] [CrossRef] [Green Version]
- Grin, Y. Comprehensive Inorganic Chemistry II; Elsevier: Oxford, UK, 2013; Volume 2, pp. 359–373. [Google Scholar]
- Bende, D.; Wagner, F.R.; Grin, Y. 8—N Rule and Chemical Bonding in Main-Group MgAgAs-Type Compounds. Inorg. Chem. 2015, 54, 3970–3978. [Google Scholar] [CrossRef]
- Evers, J. Transformation of Three-Connected Silicon in BaSi2. J. Solid State Chem. 1980, 32, 77–86. [Google Scholar] [CrossRef]
- Goebel, T.; Prots, Y.; Haarmann, F. Refinement of the crystal structure of dibarium tetrasilicide, Ba2Si4. Z. Kristallogr. NCS 2009, 224, 7–8. [Google Scholar] [CrossRef]
- Dutta, B.N. Lattice constants and thermal expansion of silicon up to 900 °C by X-ray method. Phys. Status Solidi B 1962, 2, 984–987. [Google Scholar] [CrossRef]
- Obinata, I.; Takeuchi, Y.; Kurihara, K.; Watanabe, W. Über die Legierungen des Mangans und Siliziums mit Alkali- und Erdalkalimetallen. Metallurgy 1965, 19, 21–35. [Google Scholar]
- Shi, J.; Cui, W.; Flores-Livas, J.A.; San-Miguel, A.; Botti, S.; Marques, M.A.L. Investigation of new phases in the Ba–Si phase diagram under high pressure using ab initio structural search. Phys. Chem. Chem. Phys. 2016, 18, 8108–8114. [Google Scholar] [CrossRef]
- Yamanaka, S.; Maekawa, S. Structural Evolution of the Binary System Ba-Si under High-pressure and High-temperature Conditions. Z. Naturforsch. B 2006, 61, 1493–1499. [Google Scholar] [CrossRef]
- Janzon, K.H.; Schäfer, H.; Weiss, A. Zur Kenntnis der Disilicide der Erdalkalimetalle. Z. Anorg. Allg. Chem. 1970, 372, 87–99. [Google Scholar] [CrossRef]
- Evers, J.; Oehlinger, G.; Weiss, A. Kristallstruktur von Bariumdisilicid bei hohen Drücken. Angew. Chem. 1977, 89, 673–674. [Google Scholar] [CrossRef]
- Evers, J.; Oehlinger, G.; Weiss, A. Eine neue Hochdruckphase von Bariumdisilicid. Angew. Chem. 1978, 90, 562–563. [Google Scholar] [CrossRef]
- The following idealized atomic positions have been used for the band structure computations in order to eliminate the effects of disorder: Ba1, 4e (0, 0, 0.17865); Ba2, 4d (1/2, 0, 1/4); Si1, 8f (0.3468, 0, 0); Si2, 8i (0.3324, x, 0.1014); Si3, 8i (0.2184, x, 0.4221).
- Wosylus, A.; Rosner, H.; Schnelle, W.; Schwarz, U.Z. Crystal Structure Refinement and Electronic Properties of Si(cI16). Z. Anorg. Allg. Chem. 2009, 635, 700–703. [Google Scholar] [CrossRef] [Green Version]
- Yim, W.M.; Paff, R.J. Thermal expansion of AlN, sapphire, and silicon. J. Appl. Phys. 1974, 45, 1456–1457. [Google Scholar] [CrossRef]
- Ormeci, A.; Grin, Y. Coexistence of ionic and covalent atomic interactions (bonding inhomogeneity) and thermoelectric properties of intermetallic clathrates. J. Thermoelectr. 2015, 6, 16–32. [Google Scholar]
- Grin, Y.; Fedorchuk, A.; Faria, R.J.; Wagner, F.R. Atomic Charges and Chemical Bonding in Y-Ga Compounds. Crystals 2018, 8, 99. [Google Scholar] [CrossRef]
- Kohout, M.; Wagner, F.R.; Grin, Y. Electron localization function for transition-metal compounds. Theor. Chem. Acc. 2002, 108, 150–156. [Google Scholar] [CrossRef]
- Grin, Y.; Wedig, U.; von Schnering, H.G. Hyperbolische Elektronenpaar-Struktur in RhBi4. Angew. Chem. Int. Ed. Engl. 1995, 34, 1204–1206. [Google Scholar] [CrossRef]
- Schwarz, U.; Tencé, S.; Janson, O.; Koz, C.; Krellner, C.; Burkhardt, U.; Rosner, H.; Steglich, F.; Grin, Y. CoBi3: A Binary Cobalt-Bismuth Compound and Superconductor. Angew. Chem. Int. Ed. 2013, 52, 9853–9857. [Google Scholar] [CrossRef] [PubMed]
- Guo, K.; Akselrud, L.; Bobnar, M.; Burkhardt, U.; Schmidt, M.; Zhao, J.-T.; Schwarz, U.; Grin, Y. Weak Interactions under Pressure: Hp-CuBi and Its Analogues. Angew. Chem. Int. Ed. 2017, 56, 5620–5624. [Google Scholar] [CrossRef]
- Chow, T.P.; Steckl, A.J. Refractory metal silicides: Thin-film properties and processing technology. IEEE Trans. Electron Devices 1983, 30, 1480–1497. [Google Scholar] [CrossRef]
- Flores-Livas, J.A.; Debord, R.; Botti, S.; San Miguel, A.; Marques, M.A.L.; Pailhes, S. Enhancing the Superconducting Transition Temperature of BaSi2 by Structural Tuning. Phys. Rev. Lett. 2011, 106, 087002. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Differential scanning calorimetry (DSC) curve of BaSi3 taken on heating (red curve) and cooling (blue curve) in the temperature range from 300 to 1475 K with a heating rate of 10 K/min at ambient pressure under argon atmosphere. Inset: DSC curve of BaSi3 between 300 and 625 K illustrating the onset of the exothermal decomposition at 570(5) K.
Figure 1.
Differential scanning calorimetry (DSC) curve of BaSi3 taken on heating (red curve) and cooling (blue curve) in the temperature range from 300 to 1475 K with a heating rate of 10 K/min at ambient pressure under argon atmosphere. Inset: DSC curve of BaSi3 between 300 and 625 K illustrating the onset of the exothermal decomposition at 570(5) K.

Figure 2.
Synchrotron X-ray powder diffraction pattern of BaSi3 and the result of the crystal structure refinement by the Rietveld method.
Figure 2.
Synchrotron X-ray powder diffraction pattern of BaSi3 and the result of the crystal structure refinement by the Rietveld method.

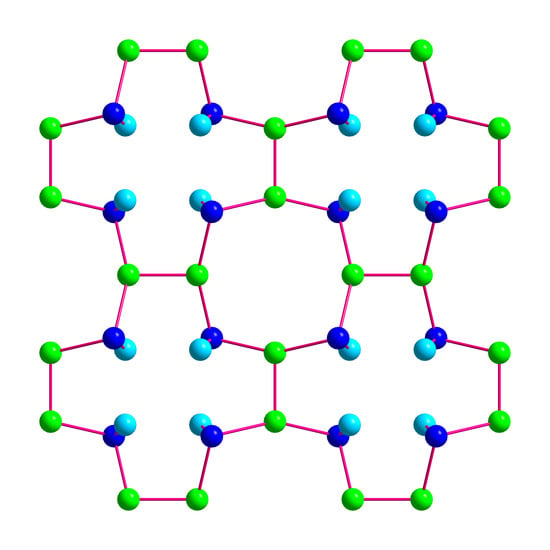
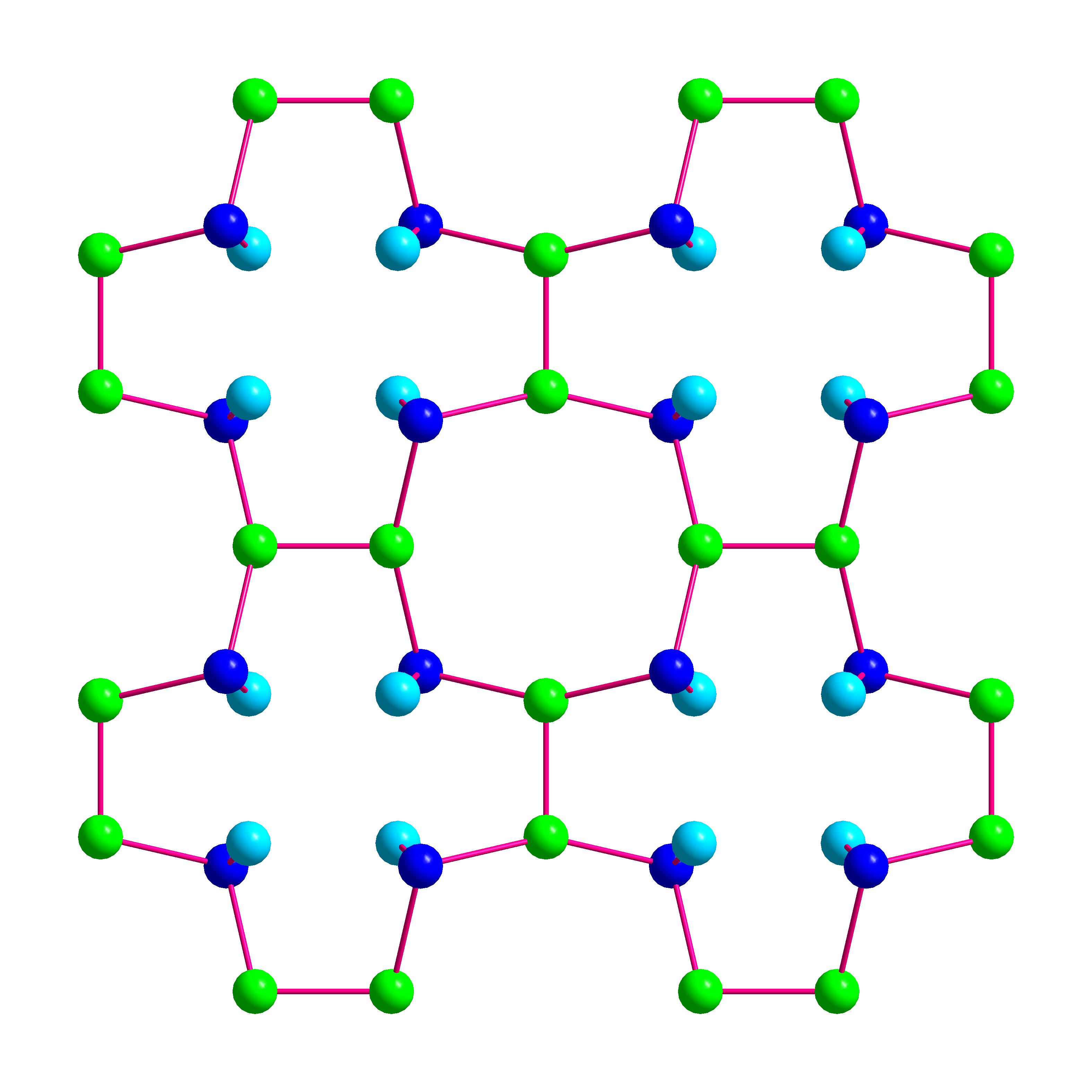
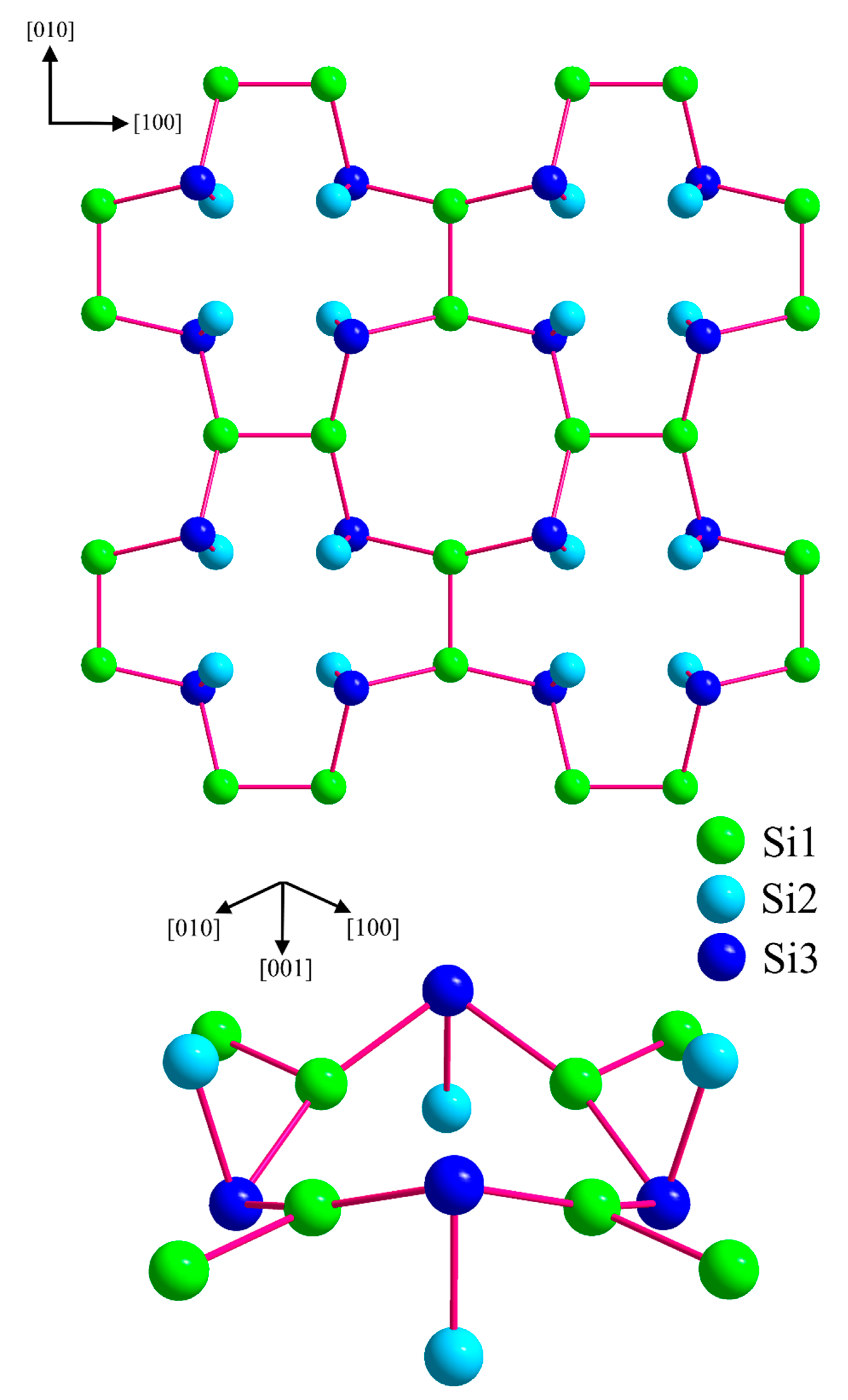
Figure 3.
Crystal structure of BaSi3. (a) Coordination polyhedrons of Ba1 and Ba2. Both positions of the disordered atom Si3 are indicated. (b) Visualization of the layer-type arrangement of BaSi3 using average positions for the disordered atoms Ba1 and Si 3. Short silicon–silicon next-neighbor distances are indicated by red lines to emphasizes the similarity of the atomic arrangement to the CaGe3-type, which is adopted by a number of related trisilicides.
Figure 3.
Crystal structure of BaSi3. (a) Coordination polyhedrons of Ba1 and Ba2. Both positions of the disordered atom Si3 are indicated. (b) Visualization of the layer-type arrangement of BaSi3 using average positions for the disordered atoms Ba1 and Si 3. Short silicon–silicon next-neighbor distances are indicated by red lines to emphasizes the similarity of the atomic arrangement to the CaGe3-type, which is adopted by a number of related trisilicides.

Figure 4.
Calculated electronic density of states (DOS) for the idealized structure model [51] of BaSi3.
Figure 4.
Calculated electronic density of states (DOS) for the idealized structure model [51] of BaSi3.

Figure 5.
The shapes of quantum theory of atoms in molecules (QTAIM) atoms and their calculated effective charges in BaSi3. The numbers are in units of elementary charge.
Figure 5.
The shapes of quantum theory of atoms in molecules (QTAIM) atoms and their calculated effective charges in BaSi3. The numbers are in units of elementary charge.

Figure 6.
Electron localizability indicator and atomic interactions in BaSi3: (left) distribution of ELI-D in the (200) plane reveals structuring of the penultimate shells of Ba1 and Ba2 as well the Si1-Si1 bonds; (right) the isosurface of ELI-D for the value of 1.5 shows Si1-Si1, Si1-Si3 and Si2-Si3 bonds as well as the five-atomic bonds Si2Ba4 and Si3Ba4.
Figure 6.
Electron localizability indicator and atomic interactions in BaSi3: (left) distribution of ELI-D in the (200) plane reveals structuring of the penultimate shells of Ba1 and Ba2 as well the Si1-Si1 bonds; (right) the isosurface of ELI-D for the value of 1.5 shows Si1-Si1, Si1-Si3 and Si2-Si3 bonds as well as the five-atomic bonds Si2Ba4 and Si3Ba4.

Figure 7.
Anionic silicon partial structure in BaSi3. (top) Covalent silicon layers with the 2c2e interactions of silicon indicated by red lines, (bottom) detail of the partial structure for visualizing the connectivity and the orientation of the (Si1)2 and the (Si2-Si3) dumbbells in more detail. Please note that the disorder of Si3 is eliminated in this idealized structure model [51].
Figure 7.
Anionic silicon partial structure in BaSi3. (top) Covalent silicon layers with the 2c2e interactions of silicon indicated by red lines, (bottom) detail of the partial structure for visualizing the connectivity and the orientation of the (Si1)2 and the (Si2-Si3) dumbbells in more detail. Please note that the disorder of Si3 is eliminated in this idealized structure model [51].

Figure 8.
Temperature-dependent electrical resistivity ρ of BaSi3 at zero-field between 2 and 305 K. Insets: Low-temperature electrical resistivity of BaGe3 between 0.11 and 2.0 K at zero-field. The normalization was performed to eliminate changes originating from different measurement geometries. The second inset shows the temperature dependence of the magnetic susceptibility χ between 1.8 and 10 K measured in a field of 0.2 mT. The subtle decrease is attributed to a small amount of superconducting impurity, but bulk superconductivity can clearly be ruled out.
Figure 8.
Temperature-dependent electrical resistivity ρ of BaSi3 at zero-field between 2 and 305 K. Insets: Low-temperature electrical resistivity of BaGe3 between 0.11 and 2.0 K at zero-field. The normalization was performed to eliminate changes originating from different measurement geometries. The second inset shows the temperature dependence of the magnetic susceptibility χ between 1.8 and 10 K measured in a field of 0.2 mT. The subtle decrease is attributed to a small amount of superconducting impurity, but bulk superconductivity can clearly be ruled out.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Data collection, structure refinement and crystallographic information for BaSi3.
| Composition | BaSi3 |
|---|---|
| Space group, Pearson symbol | I2m (no. 121), tI32 |
| Structure type | BaSi3 |
| Unit cell parameters | |
| a/Å | 7.6971(2) |
| c/Å | 12.6605(3) |
| V/Å3 | 750.07(5) |
| Formula units per unit cell, Z | 4 |
| Measurement range | 1.0° ≤ 2θ ≤ 29.0°, 0.002° step width |
| Measured points/reflections | 14000/281 |
| R(P)/R(wP) | 0.073/0.099 |
Table 2.
Wyckoff positions, site occupancy factors (SOF), relative atomic coordinates and equivalent displacement parameters Biso (in Å) for BaSi3. The values of the estimated standard deviation consider the local correlations of powder diffraction data.
Table 2.
Wyckoff positions, site occupancy factors (SOF), relative atomic coordinates and equivalent displacement parameters Biso (in Å) for BaSi3. The values of the estimated standard deviation consider the local correlations of powder diffraction data.
| Atom | Site | SOF | x/a | y/b | z/c | Biso |
|---|---|---|---|---|---|---|
| Ba1 | 8i | 0.5 | 0.0162(1) | x | 0.1785(2) | 0.67(2) |
| Ba2 | 4d | 1.0 | 1/2 | 0 | 1/4 | 0.60(2) |
| Si1 | 8f | 1.0 | 0.347(1) | 0 | 0 | 1.37(2) |
| Si2 | 8i | 1.0 | 0.332(3) | x | 0.1012(9) | 1.16(2) |
| Si3 | 16j | 0.5 | 0.2078(9) | 0.2282(9) | 0.422(1) | 1.24(2) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hübner, J.-M.; Akselrud, L.; Schnelle, W.; Burkhardt, U.; Bobnar, M.; Prots, Y.; Grin, Y.; Schwarz, U. High-Pressure Synthesis and Chemical Bonding of Barium Trisilicide BaSi3. Materials 2019, 12, 145. https://doi.org/10.3390/ma12010145
AMA Style
Hübner J-M, Akselrud L, Schnelle W, Burkhardt U, Bobnar M, Prots Y, Grin Y, Schwarz U. High-Pressure Synthesis and Chemical Bonding of Barium Trisilicide BaSi3. Materials. 2019; 12(1):145. https://doi.org/10.3390/ma12010145
Chicago/Turabian StyleHübner, Julia-Maria, Lev Akselrud, Walter Schnelle, Ulrich Burkhardt, Matej Bobnar, Yurii Prots, Yuri Grin, and Ulrich Schwarz. 2019. "High-Pressure Synthesis and Chemical Bonding of Barium Trisilicide BaSi3" Materials 12, no. 1: 145. https://doi.org/10.3390/ma12010145
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.