
Abstract
Fabry disease is a monogenic X-linked lysosomal storage disease caused by α-galactosidase A (αGalA) deficiency. Enzyme replacement therapy through administration of the missing αGalA is currently the only accepted therapeutic option. However, this treatment is connected to high costs, has ill-defined indication criteria and its efficacy is controversially discussed. Our aim was to explore the possibility of a novel targeted substrate reduction therapy for Fabry disease. Owing to the fact that αGalA-deficient humans and mice accumulate the same glycosphingolipids (i.e. globosides, galabiosylceramide and isoglobosides), αGalA-deficient mice were crossed with mice deficient in enzymes synthesizing these classes of glycosphingolipids (i.e. globotrihexosylceramide and isoglobotrihexosylceramide synthase, respectively). Functional heart and kidney tests were performed together with an extensive biochemical analysis of urine and serum in aged mice. Lysosomal storage was assessed by thin layer chromatography and electron microscopy. We showed that depletion of globosides was sufficient to fully abolish the storage of glycosphingolipids in heart, kidney and liver and was paralleled by a complete restoration of lysosomal morphology in these organs. In contrast, in dorsal root ganglia, a depletion of both globosides and isoglobosides was necessary to fully counteract the lysosomal storage. The deficiency in globosides and/or isoglobosides did not cause any adverse effects. We conclude that substrate reduction therapy through inhibition of the synthesis of globosides and isoglobosides represents a valuable therapeutic option for Fabry disease, all the more as globosides and isoglobosides seem to be dispensable.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
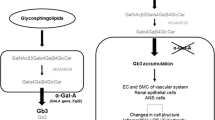
Fabry disease (Angiokeratoma corporis diffusum, OMIM 301500) was simultaneously described in 1898 by the German dermatologist Johannes Fabry and the British surgeon William Anderson (Fabry 1898; Anderson 1898). It is a monogenetic X-linked disorder characterized by different mutations of the α-galactosidase A (αGalA) gene including deletions, missense/nonsense, and frame-shift mutations or splice site defects (Brady et al. 1967; Topaloglu et al. 1999; Auray-Blais et al. 2008). The resulting decreased or absent activity of the enzyme αGalA leads to lysosomal accumulation of glycosphingolipids (GSL), the most prominent of them being globotrihexosylceramide (Gb3; Fig. 1) (Sweeley and Klionsky 1963; Hozumi et al. 1990).

Glycosphingolipid metabolism. Relevant glycosphingolipids are depicted together with the corresponding synthesis (black) and degradation (red) enzymes. Depending on the first hexose moiety added to ceramide (Cer), either galactosylceramide (GalCer) or glucosylceramide (GlcCer) is formed. GlcCer is further processed to lactosylceramide (LacCer, i.e. GalGlcCer). Through the action of further enzymes on GalCer or LacCer, individual groups of GSL are produced, from which only the first member is shown for the sake of clarity. Globotrihexosylceramide synthase (Gb3S) is responsible for the synthesis of Gb3 and galabiosylceramide (Gal 2 Cer, i.e. GalGalCer). Isoglobotrihexosylceramide synthase (iGb3S) synthesizes iGb3. α-Galactosidase A (αGalA), which catalyzes the degradation of Gb3, iGb3 and Gal2Cer, is deficient in Fabry disease. In contrast, patients with Gaucher disease suffer from GSL accumulation due to a deficiency in acid β-glucosidase (βGC). GCS glucosylceramide synthase; GlcNAc N-acetyl glucosamine; NANA N-acetylneuraminic acid; S sulfate
Fabry disease is one of the most frequent storage disorders. The incidence of the classical phenotype is estimated as 1:40,000. Recent studies, also including late-onset (non-classical) phenotypes, have reported an incidence between 1:3,100 to 1:4,600 newborns (Spada et al. 2006; Hwu et al. 2009). Random inactivation of one of the X-chromosomes in females and mutations with partially preserved enzyme activity lead to atypical or attenuated disease manifestations.
In male patients with the classical phenotype, clinical symptoms usually begin in childhood and include characteristic skin lesions (angiokeratomas), acroparesthesias, corneal opacity, and hypohidrosis. The kidney involvement manifests as proteinuria and declining kidney function which inevitably progresses to end-stage kidney disease. Cardiac manifestations include left ventricular hypertrophy, conduction abnormalities, and coronary artery disease with ensuing congestive heart failure, arrhythmias, and myocardial infarction, respectively. Transient ischemic attacks, early strokes, white matter lesions, vertigo, and hearing loss belong to further prominent, however unspecific, symptoms (reviewed in Desnick et al. 2003; Clarke 2007; Schiffmann 2009).
Currently, the only accepted treatment for Fabry disease is enzyme replacement therapy (ERT) with one of the two preparations: Agalsidase-alfa (Replagal®; Shire HGT) or agalsidase-beta (Fabrazyme®; Genzyme) (Brady 2006; Schiffmann 2009).
Although ERT is generally well tolerated and has been shown to reduce the tissue and plasma Gb3 concentration in Fabry patients, no consistent evidence of clinical efficacy could be provided (Sheppard et al. 2010; Pisani et al. 2012; Rombach et al. 2013a; El Dib et al. 2013; Tøndel et al. 2013). Due to the low prevalence of the disease and variable definitions of end-points in the available studies, therapy indications are still ill-defined—a non-negligible aspect with regard to the substantial costs of the aforementioned ERT drugs (Rombach et al. 2013b). Moreover, ERT is linked to several problems such as insufficient tissue availability (e.g., due to the blood–brain barrier) or formation of antibodies against αGalA which may have neutralizing effects or lead to infusion-associated reactions (Ohashi et al. 2008; Deegan 2012; Rombach et al. 2012; Wilcox et al. 2012).
As a monogenic disorder, Fabry disease might also be amenable to gene therapy. Although this approach has been repeatedly tested in mice, the achievement of a safe delivery of the nucleic acid to all target cells and a sustained expression of the enzyme are still the major hurdles of such therapy in humans (Jung et al. 2001; Przybylska et al. 2004; Yoshimitsu et al. 2006; Choi et al. 2010).
A different approach in the treatment of storage disorders is represented by substrate reduction therapy (SRT). In contrast to Fabry patients, individuals with Gaucher disease suffer from lysosomal storage of all GlcCer-derived GSL (Fig. 1). For the treatment of Gaucher patients, an SRT has been established and is based on the inhibition of GlcCer synthase (GCS) (Zimran 2011). This approach is also being considered for Fabry disease (Abe et al. 2000; Platt et al. 2003; Marshall et al. 2010). However, GCS inhibition depletes more GSL groups than would be needed to interfere with the storage caused by αGalA deficiency in Fabry patients (Fig. 1). As GlcCer-derived GSL play an indispensable role in numerous biological processes such as embryogenesis, central and peripheral nervous function, or epidermal skin barrier (Jennemann and Gröne 2013), their depletion may under certain circumstances be accompanied by adverse effects (Hollak et al. 2009; Machaczka et al. 2012).
We hypothesized that, for Fabry disease, an SRT by a targeted depletion of globosides and/or isoglobosides would suffice to counteract the lysosomal storage phenotype without affecting the synthesis of other GlcCer-based GSL and thus potentially lead to fewer side-effects. Our results demonstrate in vivo that interfering with the synthesis of globosides and isoglobosides fully reverted the Fabry phenotype. Therefore, an SRT by inhibition of enzymes synthesizing globosides and isoglobosides represents a valuable therapeutic option for Fabry disease and should be considered in future research.
Materials and methods
Mice
Mice deficient for Gb3S (EC 2.4.1.228, A4galttm1.1Poru) and iGb3S (EC 2.4.1.87, A3galt2tm1.1Hjg) were generated by our group and backcrossed for more than 10 generations to the C57BL/6 genetic background (Porubsky et al. 2007, 2012). Mice deficient for αGalA (EC 3.2.1.22, Glatm1Kul) were provided by Ashok Kulkarni (Ohshima et al. 1997). All strains were housed under specific pathogen-free conditions and kept under a 12/12 h light/dark cycle with access to water and regular chow ad libitum.
Urine was collected using metabolic cages with water access ad libitum. Analysis of plasma and urine samples was performed on an Hitachi 9-17E analyser (Hitachi, Frankfurt, Germany). Creatinine clearance was calculated as UCrxV24/(PCrx24x60), where UCr and PCr are creatinine concentrations in urine and plasma, respectively, and V24 is the 24-h urine volume. Peripheral blood, spleen, and thymus were analyzed by flow cytometry as detailed in Porubsky et al. (2007, 2012).
Transthoracic echocardiography
Non-invasive transthoracic echocardiography was performed in a modified setting as previously described (Malekar et al. 2010). In brief, a two-dimensional parasternal short axis view and M-mode tracings of the left ventricle were obtained with a Sonos 5500 echocardiogram (Philips, Andover, MA, USA) using a S12 transducer (12 MHz). M-mode tracings were used to measure left ventricular internal diameter (LVID) as the largest anteroposterior diameter in either diastole (LVIDd) or systole (LVIDs) and averaged from at least three consecutive cardiac cycles. Left ventricular fractional shortening was calculated as (LVIDd-LVIDs)/LVIDd and expressed as a percentage. The heart rate was calculated based on the M-mode tracings.
Isolation and analysis of GSL
Freshly harvested organs were frozen in liquid nitrogen and lyophilized. Tissues were powdered and dry weight was determined. Extraction of GSL from organs and the subsequent analysis by thin layer chromatography (TLC) were performed as described in detail in Porubsky et al. (2012); briefly, GSL were eluted from organs using CHCl3/CH3OH/H2O. In order to eliminate phospholipids and triglycerides, crude extracts were treated with 1 ml 0.1 M KOH in CH3OH at 50 °C for 4 h. After neutralization with acetic acid and evaporation of CH3OH, potassium acetate was removed from lipids via reversed phase column chromatography (RP18). Neutral and acidic (sialic acid-containing) GSL were separated by ion exchange chromatography.
For TLC, an amount corresponding to 2 mg dry organ weight was loaded. Running solvent was CHCl3/CH3OH/H2O (62.5:30:6, v/v/v) for neutral GSL and CHCl3/CH3OH/ 0.2% CaCl2 in H2O (60:35:8, v/v/v) for acidic GSL. To visualize GSL, the TLC plate was sprayed with 0.2 % orcinol in 10 % sulfuric acid and incubated 10 min at 120 °C.
Histology and immunohistochemistry
Tissue samples were fixed in 4 % phosphate buffered formaldehyde, paraffin-embedded, and cut at 4 μm. The cuts were subjected to routine staining procedures including hematoxylin and eosin stain (HE), periodic acid-Schiff (PAS), and Goldner stain. For Gb3-immunohistochemistry, cryosections were fixed with formaldehyde and probed with polyclonal chicken anti-Gb3 antibody JM06/298-1 (Betz et al. 2011) followed by alkaline phosphatase-conjugated polyclonal donkey anti-IgY secondary antibody (Jackson ImmunoResearch Europe, Suffolk, UK). Slides were scanned by Mirax Scan (Carl Zeiss, Germany). Pictures were exported using the Panoramic Viewer (3DHISTECH, Budapest, Hungary).
Electron microscopy
Organs were fixed in Karnovsky’s glutaraldehyde (2 % paraformaldehyde, 2.5 % glutaraldehyde, 0.2 M cacodylate buffer pH 7.4) and embedded in araldite (Serva, Heidelberg, Germany). Ultrathin sections were stained with lead citrate and uranyl acetate. Photographs were taken on an electron microscope (EM 910; Carl Zeiss).
Statistical analysis
Unpaired two-tailed Student’s t test was performed to compare datasets. Differences were considered significant if p < 0.05.
Results
Characterization of the gene deficient mice strains
Although αGalA-deficient mice accumulate the same GSL as humans, only a weak or non-functional organ impairment has been previously reported in αGalA-deficient mice (Ohshima et al. 1999; Yoshimitsu et al. 2006; Noben-Trauth et al. 2007; Marshall et al. 2010). Because all mice strains described in this study were backcrossed for more than 10 generations to the C57BL/6 genetic background and were strictly kept under identical conditions, we first sought to test whether, under these circumstances, any functional alterations would be detectable in αGalA-deficient mice in comparison to WT mice.
Transthoracic echocardiography did not reveal any significant difference of the left-ventricular end-diastolic diameter (LVEDD), left-ventricular end-systolic diameter (LVESD), left ventricular fractional shortening (LVFS), and ejection fraction in aged (1.5-year-old) αGalA-deficient mice as compared to WT mice (Fig. 2a–d). Similarly, the heart rate was not altered in αGalA-deficient mice (Fig. 2e). Assessment of the kidney function using metabolic cages did not show any significant changes in creatinine clearance, urine volume, proteinuria, and albuminuria in αGalA-deficient mice in comparison to WT mice (Fig. 2f–i).

Functional analysis of the heart and kidney. In aged (1.5-year-old) αGalA −/− female mice, heart and kidney function were measured. a–e The heart function was tested using transthoracic echocardiography. No significant difference of the left-ventricular end-diastolic diameter (LVEDD, a), left-ventricular end-systolic diameter (LVESD, b), left ventricular fractional shortening (c) and ejection fraction (d) could be observed between αGalA −/− and WT mice. Similarly, no difference in the heart rate could be demonstrated between αGalA-deficient and WT mice (e). Bars represent means, and whiskers standard deviations, n = 5 and 8 for WT and αGalA −/− mice, respectively. f–i The kidney function was tested based on serum creatinine and analysis of 24-h urine. No differences in the creatinine clearance (f), urine volume (g), proteinuria (h), and albuminuria (i) could be demonstrated between αGalA −/− and WT mice. Bars represent means, and whiskers standard deviations, n = 9
As further detailed in the Table 1, αGalA deficiency did not impact body and organ weights; the only exception being the kidney weight which was significantly increased in αGalA −/− females as compared to WT, αGalA −/− /Gb3S −/−, and Gb3S −/− female mice. Similarly, among all biochemical parameters tested in serum and urine, an increased cholesterol level could only be observed in αGalA –/0 male mice as compared to WT, αGalA –/0 /Gb3S −/−, and Gb3S −/− male mice (Table 1).
Mice with deficiency for globotrihexosylceramide synthase (Gb3S) and isoglobotrihexosylceramide synthase (iGb3S) have been previously reported, and the deficiency in globosides and/or isoglobosides did not impact either organ histology or any physiological function so far tested (Electronic Supplementary Material, Fig. S1; Porubsky et al. 2007, 2012).
Due to the absence of a functional organ impairment in αGalA-deficient mice with a pure genetic background, we based all further investigations on biochemical and ultrastructural analyses.
Biochemical analysis of GSL stored in αGalA-deficient mice
In αGalA-deficient humans and mice, Gb3 has been implicated as the most prominent GSL stored (Fig. 1). In order to test this in a genetic approach, αGalA-deficient mice were crossed with Gb3S −/− mice (αGalA −/− /Gb3S −/−) and tissue GSL were extracted. The analysis was focused on kidney, liver, and heart as these organs have been previously shown to have a prominent GSL accumulation in humans and mice (Hozumi et al. 1990; Ohshima et al. 1999).
TLC analysis of neutral GSL extracted from αGalA-deficient mice showed that, in kidney, liver, and heart, the most prominent accumulating GSL ran at the height of Gb3 (Fig. 3a–c). In addition, in kidneys of αGal-deficient mice, galabiosylceramide, another product of Gb3S, accumulated. In the αGalA −/− /Gb3S −/− double knockout-mice, the deficiency in Gb3S could fully abolish the GSL accumulation as detected by TLC (Fig. 3a–c). As a consequence of the Gb3S deficiency, a slight increase in its substrate LacCer could be seen in kidneys of αGalA −/− /Gb3S −/− mice (Fig. 3a–c). However, this did not led to any compensatory increase in other LacCer-derived GSL (Figs. 1, 3). No accumulation of acidic GSL occurred in αGalA-deficient mice (Fig. 3d–f).

Analysis of GSL from selected organs. Neutral (a–c) and acidic (d–f) GSL extracted from kidneys, livers, and hearts of WT, αGalA −/−, and αGalA −/− /Gb3S −/− mice were analyzed by TLC. In all three organs, αGalA deficiency resulted in accumulation of a neutral GSL which ran at the height of Gb3 and was absent in the αGalA −/− /Gb3S −/− double knockout thus confirming its identity as Gb3 (a–c). Accumulation of galabiosylceramide (Gal 2 Cer) contributed to the storage in kidneys of αGalA −/− mice (a), but was—in agreement with being a product of Gb3S—absent in αGalA −/− /Gb3S −/− kidneys. Gb3S deficiency resulted in a slight accumulation of its substrate lactosylceramide (LacCer) in kidneys (a). Among the acidic GSL (d—f), no accumulation was observed in αGalA −/− mice. Some GSL are represented by multiple bands due to their heterogeneous composition of fatty acids resulting in different running properties. Orcinol staining. Std. GSL standard
Impact of Gb3S deficiency on the storage phenotype in αGalA-deficient mice
To corroborate the TLC findings with histology, immunohistochemistry with anti-Gb3 antibodies was performed on kidney and liver tissue. In WT kidneys, a strong Gb3 expression could be observed in collecting ducts (Fig. 4a). In αGalA-deficient mice, the Gb3 synthesis became apparent due to the absence of its catabolism also in other nephron segments (Fig. 4b). In contrast, glomerular cells stained negative for Gb3 in WT mice as well as in αGalA-deficient mice (data not shown). In congruence with the TLC analysis, livers showed no Gb3 expression upon immunohistochemical investigation in WT mice but its accumulation was present in αGalA-deficient animals (Fig. 4d, e). Gb3S deficiency abolished the accumulation of globosides observed in organs of αGalA-deficient mice (Fig. 4c, f).

Immunohistochemical analysis of the Gb3 storage. Immunohistochemistry was performed using Gb3-antibodies in kidneys (a–c) and livers (d–f) of WT, αGalA −/−, and αGalA −/− /Gb3S −/− mice. Gb3 was prominently present in renal collecting ducts of WT mice but also emerged in other nephron segments in αGalA deficiency. Similarly, in the liver, Gb3 also accumulated in αGalA −/− mice. Gb3S deficiency abolished this storage in αGalA −/− /Gb3S −/− mice. Scale bars (a–c) 0.5 mm, (d–f) 0.1 mm
Furthermore, organs were subjected to ultrastructural analysis, which represents the decisive method for visualization of lysosomal storage phenomena. In αGalA-deficient mice, the storage could be documented as concentric and lamellar lysosomal inclusions (Fig. 5b, e, h) which are also characteristic for the human disease. In line with the immunohistochemical findings, an extensive accumulation could be documented in renal tubular epithelial cells in αGalA-deficient mice (Fig. 5b, e). In contrast, podocytes and renal endothelial cells of αGalA-deficient mice showed a regular ultrastructure. Singular podocytes alone showed a slight lysosomal enlargement and lamellar structures resembling αGalA deficiency (data not shown).

Ultrastructural analysis of selected organs. Organs of WT, αGalA −/−, and αGalA −/− /Gb3S −/− mice were analyzed by electron microscopy for the presence of ultrastructural correlates of a lysosomal storage disease. a–f In kidneys of αGalA −/− mice, numerous enlarged lysosomes with characteristic concentric lamellar inclusions were present in tubular epithelial cells (black arrow). Depletion of globosides fully restored a normal lysosomal morphology in αGalA −/− /Gb3S −/− mice. g–i The storage phenotype in the liver is exemplified by Ito cells, which can be identified in the Disse’s (perisinusoid) space (*endothelial cells). αGalA deficiency resulted in a distorted lysosomal architecture (white arrow) in αGalA −/− mice but not in the absence of Gb3S-activity (i.e. in the αGalA −/− /Gb3S −/− mice). Stain: lead citrate/uranyl acetate. Scale bars(a–c) 5 μm, (d–f) 2 μm, (g–i) 0.5 μm
Electron microscopy revealed the lysosomal storage also in Kupffer and Ito cells in livers of αGalA-deficient mice (Fig. 5h). In hearts of αGalA-deficient mice, endothelial and interstitial cells were affected by the storage (Electronic Supplementary Material, Fig. S2).
In αGalA −/− /Gb3S −/− mice, the elimination of globosides restored a normal lysosomal morphology in kidney, liver and heart and resulted in an ultrastructural appearance indistinguishable from WT mice (Fig. 5c, f, i; Electronic Supplementary Material, Fig. S2). Thus, Gb3S deficiency was sufficient to abolish the lysosomal storage associated with αGalA deficiency in these organs, yet without causing any developmental or histological abnormalities and without interfering with organ function (Table 1 and Porubsky et al. 2007, 2012).
Contribution of isoglobosides to the storage phenotype in dorsal root ganglia
A comprehensive ultrastructural analysis of other structures revealed that—in contrast to the previously mentioned organs—in dorsal root ganglia (DRG) of αGalA −/− /Gb3S −/− mice, a lysosomal storage was still present (Fig. 6). Taking into account that iGb3 is synthesized in DRG (Speak et al. 2007), we analyzed DRG of αGalA −/− /iGb3S −/− mice. However, the sole deficiency in iGb3S did not suffice to counteract the storage phenotype associated with αGalA deficiency in DRG. Only a simultaneous depletion of both globosides and isoglobosides (i.e. in the αGalA −/− /Gb3 −/− /iGb3 −/− mouse) was able to abolish the lysosomal storage and restore a normal ultrastructural morphology in the DRG (Fig. 6h, j).

Ultrastructural analysis of dorsal root ganglia. Dorsal root ganglia were analyzed by electron microscopy. DRG of αGalA-deficient mice showed numerous enlarged lysosomes filled with lamellar material. However, in contrast to other organs, a restoration of a normal lysosomal morphology could not be achieved by depletion of globosides (αGalA −/− /Gb3S −/−). Similarly, depletion of isoglobosides (αGalA −/− /iGb3S −/−) also did not lead to a normalization of the lysosomal morphology. This could be achieved only by a simultaneous depletion of both globosides and isoglobosides (i.e. in the αGalA −/− /Gb3S −/− /iGb3S −/− mouse). Stain: lead citrate/uranyl acetate. Scale bars (a–c, g, h) 2 μm, (d–f, i, j) 1 μm
Discussion
These results demonstrate that, in αGalA deficiency, depletion of globosides was sufficient to fully revert the storage phenotype in the most prominently affected organs like heart, liver, and kidney (Figs. 3, 4, 5, 6). To achieve a complete restoration of the lysosomal morphology in DRG, which also produce isoglobosides, the simultaneous depletion of both globosides and isoglobosides was necessary (Fig. 6). The latter finding is supported by a previous investigation, which documented the synthesis of iGb3 only in the DRG but not in other murine organs (Speak et al. 2007).
The murine model of Fabry disease used in this study shows—in congruence with the human disease—a complete lack of lysosomal αGalA activity, which results in an aberrant lysosomal ultrastructure and function (Ohshima et al. 1997; Ohashi et al. 2008; Porubsky et al. 2012). However, in contrast to Fabry patients, an effect on the function of heart, kidney, and brain is completely absent or only very mild, and the life span is normal in αGalA-deficient mice. In particular, no ophthalmologic manifestations (Ohshima et al. 1999) and no hearing loss (Noben-Trauth et al. 2007) could be demonstrated, although some studies have reported slight sensorimotor alterations (Rodrigues et al. 2009; Marshall et al. 2010) and a marginal myocardial affection (Yoshimitsu et al. 2006; Rozenfeld et al. 2011; Nguyen Dinh Cat et al. 2012). The latter could not be verified in our colony, possibly due to the extensive backcrossing towards the reference C57BL/6 strain and a strict breeding of all strains under identical conditions (Fig. 2). Also in line with previous investigations (Ohshima et al. 1999), we could not detect any decline in kidney function (measured as creatinine clearance) and no proteinuria or microalbuminuria in aged αGalA-deficient mice (Fig. 2). The increased kidney weight in αGalA −/− females as compared to WT, αGalA −/− /Gb3S −/−, and Gb3S −/− mice (Table 1) did not show any correlates upon light microscopy (data not shown). Similarly, the elevated serum cholesterol in male αGalA –/0 mice (Table 1) was not reflected by any light microscopic changes in the liver or any other organ (including heart; data not shown).
The aforementioned absent or very slight functional alterations in the murine model of Fabry disease contrast with the extensive GSL accumulation and the amply documented derangement of lysosomal morphology in the organs of these mice (Figs. 3, 4, 5, 6). The paucity of functional correlates in this mouse model is probably due to the fact that the cells which determine the organ affection and morbidity in humans (e.g., podocytes in the kidney or endothelial cells in the heart and brain) do not express sufficient amounts of Gb3S in mice and are thus not affected by the storage to an extent which would allow the development of similar symptoms. This explanation is supported by a recent report which showed that αGalA-deficient mice were symptomatic when crossbred with transgenic mice ubiquitously expressing Gb3S (Taguchi et al. 2013). αGalA-deficient mice and humans show identical GSL storage patterns with globosides constituting the vast majority of the accumulating GSL (Sweeley and Klionsky 1963; Hozumi et al. 1990; Ohshima et al. 1999). Therefore, although the αGalA-deficient mouse model is not suited for functional studies, it represents a useful tool to investigate biochemical and ultrastructural alterations and possible therapeutic approaches in vivo. In our study, we did not determine the levels of lyso-Gb3, which has been postulated as a marker of Fabry disease (Aerts et al. 2008; Togawa et al. 2010), as it can be anticipated that, in the αGalA −/− /Gb3S −/− mouse, the abolishment of the synthesis of Gb3 will curtail any production of lyso-Gb3 (Fig. 3).
To date, the only accepted therapy for Fabry disease is ERT. Besides its high costs and still unproven efficacy, the most prominent problem of the intravenously administered enzyme is its bioavailability in target organs. In this regard, an approach through SRT seems to be advantageous because tissue availability of small inhibitors can be optimized by molecular engineering. SRT through inhibition of GCS (Fig. 1) seems to be possible as a therapeutic option for αGalA deficiency (Abe et al. 2000; Platt et al. 2003). However, such an approach also results in depletion of other GSL groups, which do not contribute to the storage in αGalA deficiency. Moreover, GSC-inhibition does not interfere with the synthesis of galabiosylceramide in the kidney (Fig. 1) and thus cannot abolish its accumulation in Fabry disease. Although it is unclear to which extent the accumulation of galabiosylceramide itself contributes to the renal phenotype of Fabry disease, inhibition of the Gb3S would—in contrast to the inhibition of GCS—eliminate the accumulation not only of globosides but also of galabiosylceramide.
It could be demonstrated that deficiency in globosides is protective upon exposure to Shigatoxins (Okuda et al. 2006; own unpublished data); however, the physiological function of globosides and isoglobosides remains elusive as no derangements of physiological functions have so far been observed in mice deficient for Gb3S and/or iGb3S (Table 1; Electronic Supplementary Material, Fig.S1; Porubsky et al. 2007, 2012).
In summary, our study explored the possibility of a targeted SRT for Fabry disease through the depletion of globosides and isoglobosides, which both seem to be dispensable for all biological functions tested so far (Table 1; Porubsky et al. 2007, 2012). We show that interference with the Gb3S activity abolishes the GSL storage and normalizes lysosomal morphology in most organs. For a complete elimination of storage phenomena in DRG, isoglobosides have to be depleted in addition to globosides. We are convinced that a pharmacologic inhibition of Gb3S and iGb3S represents a potentially successful SRT for Fabry disease. Experiments are in progress to identify specific inhibitors of Gb3S and iGb3S.
References
Abe A, Gregory S, Lee L, Killen PD, Brady RO, Kulkarni A, Shayman JA (2000) Reduction of globotriaosylceramide in Fabry disease mice by substrate deprivation. J Clin Invest 105:1563–1571
Aerts JM, Groener JE, Kuiper S, Donker-Koopman WE, Strijland A, Ottenhoff R, van Roomen C, Mirzaian M, Wijburg FA, Linthorst GE, Vedder AC, Rombach SM, Cox-Brinkman J, Somerharju P, Boot RG, Hollak CE, Brady RO, Poorthuis BJ (2008) Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc Natl Acad Sci USA 105:2812–2817
Anderson W (1898) A case of “angeio-keratoma”. Br J Dermatol 10:113–117
Auray-Blais C, Cyr D, Ntwari A, West ML, Cox-Brinkman J, Bichet DG, Germain DP, Laframboise R, Melançon SB, Stockley T, Clarke JTR, Drouin R (2008) Urinary globotriaosylceramide excretion correlates with the genotype in children and adults with Fabry disease. Mol Genet Metab 93:331–340
Betz J, Bielaszewska M, Thies A, Humpf HU, Dreisewerd K, Karch H, Kim KS, Friedrich AW, Muthing J (2011) Shiga toxin glycosphingolipid receptors in microvascular and macrovascular endothelial cells: differential association with membrane lipid raft microdomains. J Lipid Res 52:618–634
Brady RO (2006) Enzyme Replacement for Lysosomal Diseases. Annu Rev Med 57:283–296
Brady RO, Gal AE, Bradley RM, Martensson E, Warshaw AL, Laster L (1967) Enzymatic Defect in Fabry’s Disease. N Engl J Med 276:1163–1167
Choi J, Lee MH, Park H, Jung S (2010) Characterization of Fabry mice treated with recombinant adeno-associated virus 2/8-mediated gene transfer. J Biomed Sci 17:26
Clarke JTR (2007) Narrative review: Fabry disease. Ann Intern Med 146:425–433
Deegan P (2012) Fabry disease, enzyme replacement therapy and the significance of antibody responses. J Inherit Metab Dis 35:227–243
Desnick RJ, Brady R, Barranger J, Collins AJ, Germain DP, Goldman M, Grabowski G, Packman S, Wilcox WR (2003) Fabry Disease, an Under-Recognized Multisystemic Disorder: Expert Recommendations for Diagnosis, Management, and Enzyme Replacement Therapy. Ann Intern Med 138:338–346
El Dib RP, Nascimento P, Pastores GM (2013) Enzyme replacement therapy for Anderson-Fabry disease. Cochrane Database Syst Rev 2, CD006663
Fabry J (1898) Ein Beitrag zur Kenntniss der Purpura haemorrhagica nodularis (Purpura papulosa haemorrhagica Hebrae) [A contribution to knowledge on the purpura haemorrhagica nodularis (purpura papulosa haemorrhagica Hebrae)]. Arch Dermatol Syph 43:187–200
Hollak CEM, Hughes D, van Schaik IN, Schwierin B, Bembi B (2009) Miglustat (Zavesca®) in type 1 Gaucher disease: 5-year results of a post-authorisation safety surveillance programme. Pharmacoepidemiol Drug Saf 18:770–777
Hozumi I, Nishizawa M, Ariga T, Miyatake T (1990) Biochemical and clinical analysis of accumulated glycolipids in symptomatic heterozygotes of angiokeratoma corporis diffusum (Fabry’s disease) in comparison with hemizygotes. J Lipid Res 31:335–340
Hwu WL, Chien YH, Lee NC, Chiang SC, Dobrovolny R, Huang AC, Yeh HY, Chao MC, Lin SJ, Kitagawa T, Desnick RJ, Hsu LW (2009) Newborn screening for Fabry disease in Taiwan reveals a high incidence of the later-onset GLA mutation c.936 + 919G > A (IVS4 + 919G > A). Hum Mutat 30:1397–1405
Jennemann R, Gröne H-J (2013) Cell-specific in vivo functions of glycosphingolipids: Lessons from genetic deletions of enzymes involved in glycosphingolipid synthesis. Prog Lipid Res 52:231–248
Jung S-C, Han IP, Limaye A, Xu R, Gelderman MP, Zerfas P, Tirumalai K, Murray GJ, During MJ, Brady RO, Qasba P (2001) Adeno-associated viral vector-mediated gene transfer results in long-term enzymatic and functional correction in multiple organs of Fabry mice. Proc Natl Acad Sci USA 98:2676–2681
Machaczka M, Hast R, Dahlman I, Lerner R, Klimkowska M, Engvall M, Hägglund H (2012) Substrate reduction therapy with miglustat for type 1 Gaucher disease: a retrospective analysis from a single institution. Ups J Med Sci 117:28–34
Malekar P, Hagenmueller M, Anyanwu A, Buss S, Streit MR, Weiss CS, Wolf D, Riffel J, Bauer A, Katus HA, Hardt SE (2010) Wnt signaling is critical for maladaptive cardiac hypertrophy and accelerates myocardial remodeling. Hypertension 55:939–945
Marshall J, Ashe KM, Bangari D, McEachern K, Chuang W-L, Pacheco J, Copeland DP, Desnick RJ, Shayman JA, Scheule RK, Cheng SH (2010) Substrate Reduction Augments the Efficacy of Enzyme Therapy in a Mouse Model of Fabry Disease. PLoS ONE 5:e15033
Nguyen Dinh Cat A, Escoubet B, Agrapart V, Griol-Charhbili V, Schoeb T, Feng W, Jaimes E, Warnock DG, Jaisser F (2012) Cardiomyopathy and Response to Enzyme Replacement Therapy in a Male Mouse Model for Fabry Disease. PLoS ONE 7:e33743
Noben-Trauth K, Neely H, Brady RO (2007) Normal hearing in α-galactosidase A-deficient mice, the mouse model for Fabry disease. Hear Res 234:10–14
Ohashi T, Iizuka S, Ida H, Eto Y (2008) Reduced α-Gal A enzyme activity in Fabry fibroblast cells and Fabry mice tissues induced by serum from antibody positive patients with Fabry disease. Mol Genet Metab 94:313–318
Ohshima T, Murray GJ, Swaim WD, Longenecker G, Quirk JM, Cardarelli CO, Sugimoto Y, Pastan I, Gottesman MM, Brady RO, Kulkarni AB (1997) alpha-Galactosidase A deficient mice: a model of Fabry disease. Proc Natl Acad Sci USA 94:2540–2544
Ohshima T, Schiffmann R, Murray GJ, Kopp J, Quirk JM, Stahl S, Chan CC, Zerfas P, Tao-Cheng JH, Ward JM, Brady RO, Kulkarni AB (1999) Aging accentuates and bone marrow transplantation ameliorates metabolic defects in Fabry disease mice. Proc Natl Acad Sci USA 96:6423–6427
Okuda T, Tokuda N, Numata S, Ito M, Ohta M, Kawamura K, Wiels J, Urano T, Tajima O, Furukawa KK (2006) Targeted disruption of Gb3/CD77 synthase gene resulted in the complete deletion of globo-series glycosphingolipids and loss of sensitivity to verotoxins. J Biol Chem 281:10230–10235
Pisani A, Visciano B, Roux GD, Sabbatini M, Porto C, Parenti G, Imbriaco M (2012) Enzyme replacement therapy in patients with Fabry disease: State of the art and review of the literature. Mol Genet Metab 107:267–275
Platt FM, Jeyakumar M, Andersson U, Heare T, Dwek R a, Butters TD (2003) Substrate reduction therapy in mouse models of the glycosphingolipidoses. Philos Trans R Soc Lond B 358:947–954
Porubsky S, Speak AO, Luckow B, Cerundolo V, Platt FM, Gröne H-J (2007) Normal development and function of invariant natural killer T cells in mice with isoglobotrihexosylceramide (iGb3) deficiency. Proc Natl Acad Sci USA 104:5977–5982
Porubsky S, Speak AO, Salio M, Jennemann R, Bonrouhi M, Zafarulla R, Singh Y, Dyson J, Luckow B, Lehuen A, Malle E, Muthing J, Platt FM, Cerundolo V, Grone HJ (2012) Globosides but not isoglobosides can impact the development of invariant NKT cells and their interaction with dendritic cells. J Immunol 189:3007–3017
Przybylska M, Wu I-H, Zhao H, Ziegler RJ, Tousignant JD, Desnick RJ, Scheule RK, Cheng SH, Yew NS (2004) Partial correction of the alpha-galactosidase A deficiency and reduction of glycolipid storage in Fabry mice using synthetic vectors. J Gene Med 6:85–92
Rodrigues LG, Ferraz MJ, Rodrigues D, Pais-Vieira M, Lima D, Brady RO, Sousa MM, Sá-Miranda MC (2009) Neurophysiological, behavioral and morphological abnormalities in the Fabry knockout mice. Neurobiol Dis 33:48–56
Rombach SM, Aerts JMFG, Poorthuis BJHM, Groener JEM, Donker-Koopman W, Hendriks E, Mirzaian M, Kuiper S, Wijburg F a, Hollak CEM, Linthorst GE (2012) Long-term effect of antibodies against infused alpha-galactosidase A in Fabry disease on plasma and urinary (lyso)Gb3 reduction and treatment outcome. PLoS ONE 7:e47805
Rombach S, Smid B, Bouwman M, Linthorst G, Dijkgraaf MGW, Hollak CEM (2013a) Long term enzyme replacement therapy for Fabry disease: effectiveness on kidney, heart and brain. Orphanet J Rare Dis 8:47
Rombach SM, Hollak CEM, Linthorst GE, Dijkgraaf MGW (2013b) Cost-effectiveness of enzyme replacement therapy for Fabry disease. Orphanet J Rare Dis 8:29
Rozenfeld PA, Fritz M, Blanco P, Gonzalez P, Rinaldi GJ (2011) Myocardial Alterations in the Murine Model of Fabry Disease Can Be Reversed by Enzyme Replacement Therapy. Can J Cardiol 27:339–345
Schiffmann R (2009) Fabry disease. Pharmacol Ther 122:65–67
Sheppard MN, Cane P, Florio R, Kavantzas N, Close L, Shah J, Lee P, Elliott P (2010) A detailed pathologic examination of heart tissue from three older patients with Anderson–Fabry disease on enzyme replacement therapy. Cardiovasc Pathol 19:293–301
Spada M, Pagliardini S, Yasuda M, Tukel T, Thiagarajan G, Sakuraba H, Ponzone A, Desnick RJ (2006) High incidence of later-onset fabry disease revealed by newborn screening. Am J Hum Genet 79:31–40
Speak AO, Salio M, Neville DCA, Fontaine J, Priestman DA, Platt N, Heare T, Butters TD, Dwek RA, Trottein F, Exley MA, Cerundolo V, Platt FM (2007) Implications for invariant natural killer T cell ligands due to the restricted presence of isoglobotrihexosylceramide in mammals. Proc Natl Acad Sci USA 104:5971–5976
Sweeley CC, Klionsky B (1963) Fabry’s Disease: Classification as a Sphingolipidosis and Partial Characterization of a Novel Glycolipid. J Biol Chem 238:PC3148–PC3150
Taguchi A, Maruyama H, Nameta M, Yamamoto T, Matsuda J, Kulkarni AB, Yoshioka H, Ishii S (2013) A symptomatic Fabry disease mouse model generated by inducing globotriaosylceramide synthesis. Biochem J 456:373–383
Togawa T, Kodama T, Suzuki T, Sugawara K, Tsukimura T, Ohashi T, Ishige N, Suzuki K, Kitagawa T, Sakuraba H (2010) Plasma globotriaosylsphingosine as a biomarker of Fabry disease. Mol Genet Metab 100:257–261
Tøndel C, Bostad L, Larsen KK, Hirth A, Vikse BE, Houge G, Svarstad E (2013) Agalsidase benefits renal histology in young patients with Fabry disease. J Am Soc Nephrol 24:137–148
Topaloglu AK, Ashley GA, Tong B, Shabbeer J, Astrin KH, Eng CM, Desnick RJ (1999) Twenty novel mutations in the alpha-galactosidase A gene causing Fabry disease. Mol Med 5:806–811
Wilcox WR, Linthorst GE, Germain DP, Feldt-Rasmussen U, Waldek S, Richards SM, Beitner-Johnson D, Cizmarik M, Cole JA, Kingma W, Warnock DG (2012) Anti-α-galactosidase A antibody response to agalsidase beta treatment: Data from the Fabry Registry. Mol Genet Metab 105:443–449
Yoshimitsu M, Higuchi K, Dawood F, Rasaiah VI, Ayach B, Chen M, Liu P, Medin JA (2006) Correction of cardiac abnormalities in fabry mice by direct intraventricular injection of a recombinant lentiviral vector that engineers expression of alpha-galactosidase A. Circ J 70:1503–1508
Zimran A (2011) How I treat Gaucher disease. Blood 118:1463–1471
Acknowledgements
We thank M. Bonrouhi and S. Kaden for excellent technical assistance (both at the German Cancer Research Center, Heidelberg, Germany) and A. Kulkarni (National Institutes of Health, Bethesda, MD, USA) for providing α-galactosidase A-deficient mice. We are grateful to J. Müthing (University of Munster, Germany) for the polyclonal anti-Gb3 antibody.
This work was supported by a grant from the Deutsche Forschungsgemeinschaft to H.-J. Gröne and S. Porubsky (SFB 938). L. Lehmann is recipient of the HRCMM (Heidelberg Research Center for Molecular Medicine) Career Development Fellowship.
Disclosure
All authors declare that they do not have any conflicting financial interests.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Electronic Supplementary Material, Figure S1
(PDF 200 kb)
Electronic Supplementary Material, Figure S2
(PDF 131 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Porubsky, S., Jennemann, R., Lehmann, L. et al. Depletion of globosides and isoglobosides fully reverts the morphologic phenotype of Fabry disease.. Cell Tissue Res 358, 217–227 (2014). https://doi.org/10.1007/s00441-014-1922-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-014-1922-9