
Abstract
Prader-Willi syndrome (PWS) is a neurodevelopmental disorder associated with abnormalities of chromosome 15q11q13. The majority of cases result either from a deletion approximately 4 Mb in size, affecting chromosome 15 of paternal origin or from UPD(15)mat; these account for ~70 and ~20–25% of PWS cases, respectively. In the remaining 3–5% of PWS cases where neither the deletion nor UPD is detectable, PWS is thought to be caused either by a defect in the imprinting centre resulting in a failure to reset the paternally inherited chromosome 15 derived from the paternal grandmother or, very occasionally, from a balanced translocation involving a breakpoint in 15q11q13. Nine probands with a firm clinical diagnosis of PWS but who had neither a typical deletion in the PWS region nor UPD(15)mat were investigated for inactivating mutations in 11 genes located in the PWS region, including SNURF and SNRPN, which are associated with the imprinting centre. Other genes studied for mutations included MKRN3, NDN, IPW, HBII-85, HBII-13, HBII-436, HBII-438a, PAR1 and PAR5. A possibly inactivating mutation in the SNRPN minimal promoter region was identified. No other inactivating mutations were found in the remainder of our panel of PWS subjects with atypical genetics. Expression levels of several of the candidate genes for PWS were also investigated in this series of probands. The results indicate that PWS may result from a stochastic partial inactivation of important genes.
Similar content being viewed by others
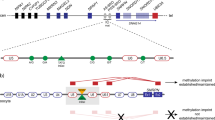


Introduction
Prader-Willi syndrome (PWS) (MIM176270) is a complex neurodevelopmental disorder affecting ~1/22,000 live births but with a population prevalence of ~1/54,000 (Whittington et al. 2001). Clinically, PWS is characterised by two phases. In infancy there is a failure to thrive, with muscle hypotonia, genital hypoplasia, respiratory problems and feeding difficulties (Holm et al. 1993; Cassidy 1997). From as early as 2 years of age, an altered phenotype becomes apparent with evidence of mild developmental delay and learning disabilities and the onset of severe over-eating behaviour resulting from an abnormal satiety response to food intake (Holland et al. 1995). This can lead to severe obesity if not controlled. Other later-phase phenotypic characteristics include short stature, small hands and feet and significant behavioural problems. A points system of eight major criteria (1 point each) and 11 minor criteria (1/2 point each) has been devised to aid in the diagnosis of PWS. A firm clinical diagnosis of PWS must reach a score of at least 8 points including at least four of the major and several of the minor criteria as addressed by Holm et al. (1993).
In a population-based study of PWS (Whittington et al. 2001), it was found that the presence of five ‘core’ clinical criteria (floppy baby, weak cry or inactivity, poor suck, feeding difficulties, and hypogonadism) was necessary, but not sufficient, for a positive genetic diagnosis (Whittington et al. 2002). In this study, two people were identified who fulfilled the clinical criteria (Holm et al. 1993) for PWS but genetically showed a normal methylation pattern at the SNURF/SNRPN exon 1 region, with concomitant expression of SNRPN. One of these individuals was heterozygous at microsatellite loci spanning the whole PWS region, while the second person was found, by two independent genetics laboratories, to have a small paternally derived deletion in q12, encompassing D15S9 (MKRN3) and D15S543 but not extending as far as D15S541.
In a further, UK-wide study of psychosis in PWS (Soni 2006), another person was found with the five ‘core’ clinical criteria, but with normal genetics. Could these atypical people possibly have a mutation in, or a deletion of, a single maternally imprinted gene that would cause PWS? A hypothetical model of a single gene cause of PWS has been put forward (Holland et al. 2003) although it is contrary to mainstream opinion. However, the region thought to contain the ‘PWS genes’ has been shrinking over the past decade. If this single gene hypothesis were true, a logical starting point for the ‘PWS gene’ would be within the small deletion in the second case referred to above.
PWS is caused by the absence of expression of a cluster of paternally expressed genes located at 15q11q13, usually achieved by an interstitial paternal chromosomal deletion, UPD(15)mat or defects in the imprinting centre (IC) (Nicholls and Knepper 2001). The ~4-Mb common deletion that accounts for 70% of PWS cases (Nicholls and Knepper 2001; Christian et al. 1995; Amos-Landgraf et al. 1999) spans the 2-Mb imprinted domain and also several non-imprinted genes. To date, there are at least 11 known paternally expressed genes in the region with potential involvement in the etiology of PWS. These include MAGEL2, a related MAGE family member coding for the protein MAGE-like 2, of unknown function but with numerous potential nuclear localisation signals that is highly expressed in the hypothalamus and other regions of the brain (Boccaccio et al. 1999; Lee et al. 2000) and NDN encoding NECDIN, a MAGE protein family member proposed to act as a neuronal growth suppressor and anti-apoptotic protein in post-mitotic neurons (Macdonald and Wevrick 1997; Jay et al. 1997; Pagliardini et al. 2005).
Another known gene, MKRN3, encodes a putative ribonucleoprotein, the makorin ring finger protein 3 (Jong et al. 1999). The complex IC-SNURF/SNRPN-snoRNA polycistronic locus encodes several functions including a cis-acting regulatory region, two nuclear proteins (SNURF and SNRPN) (Glenn et al. 1996; Gray et al. 1999), five classes of box C/D small nucleolar RNAs (snoRNAs) (Cavaille et al. 2000; de los Santos et al. 2000; Runte et al. 2001) and a UBE3A-AS antisense transcript (Rougeulle et al. 1998; Runte et al. 2001; Landers et al. 2004). SNURF/SNRPN encodes an arginine-rich polypeptide, small ribonucleoprotein N, whose putative function includes RNA binding (Gray et al. 1999). The snoRNAs in the PWS region lack the usual rRNA complementarity and may target cellular mRNAs for methylation or alternative splicing (Cavaille et al. 2000; de los Santos et al. 2000; Runte et al. 2001). HBII-85, a member of the snoRNA family, has been shown to be highly conserved in rodents, and Gallagher et al. (2002) showed that a 121-kb region thought to be critical in PWS contained both HBII-85 and HBII-438, suggesting that these genes may play a major role in the PWS phenotype.
Angelman syndrome (AS) is clinically distinct from PWS, although it also results from abnormalities in the 15q11q13 region, but here the deletion is maternally inherited and the UPD is paternally derived (Nicholls and Knepper 2001). AS therefore results from the inactivity of maternally, rather than paternally, inherited genes in 15q11q13. About 20% of the people with AS carry a causative mutation in the UBE3A gene, located in the maternally active but paternally imprinted part of the bipartite PWS/AS region. The UBE3A gene was not originally considered to be a candidate gene for AS, as it is not imprinted in blood lymphocytes. Only when the gene was found to be paternally imprinted in certain regions of the brain were mutations searched for and identified as associated with AS (Kishino et al. 1997; Rougeulle et al. 1997). To date, unlike AS, no single gene mutation has been implicated in the etiology of PWS although candidate genes have been proposed.
The human SNRPN minimal promoter region (SMP) upstream of the SNURF transcript has been hypothesized as a likely candidate where a mutation could inactivate the SNRPN gene and also affect subsequent downstream expression. The SMP region includes 71 bp of upstream sequence and the first 51 bp of SNURF/SNRPN exon 1 (Finberg et al. 2003).
This work describes a search for mutations in imprinted genes in the PWS region of chromosome 15 that might have a causative role in the syndrome combined with a study of expression levels in some of these candidate genes. Starting with the three individuals described above, the cohort was expanded to include further individuals with unusual or possibly informative genotypes.
Materials and methods
Participants
The probands included in this study were a subset from the large extended population study involving approximately a third of the people with PWS resident in the UK (Whittington et al. 2001; Soni 2006). On entry into the study, the participants were clinically and psychologically assessed, and blood samples were taken and sent to the genetics laboratory where both RNA and DNA were extracted by routine methods.
Molecular diagnosis of PWS
The absence of a paternally inherited non-methylated band at the SNURF/SNRPN locus after treatment of DNA samples with bisulphite followed by methylation-specific PCR was taken as laboratory confirmation of the diagnosis of PWS (Kubota et al. 1997; Zeschnigk et al. 1997). Confirmation of the molecular diagnosis was also obtained by a lack of expression of SNRPN (Wevrick and Francke 1996).
Microsatellite analysis
Differentiation between participants who carried a deletion in 15q11q13 and those with UPD(15)mat was made by detection of alleles at a combination of 14 microsatellite loci lying within the common deletion and two lying distal to the common breakpoint BP3 within the HERC2 gene (Chai et al. 2003). Some loci were studied by incorporation of 32P-[dCTP] followed by autoradiography, and others by the use of fluorescence-tagged primers and Genescan software (Applied Biosciences). When more densely packed markers were needed, the tool “repeat master” (ensembl.org) was used to identify further polymorphic CA repeat sequences. The microsatellite markers so obtained are identified by the contig from which they were derived.
Where possible, confirmation of the genetic subtype was obtained from parental DNA. All of the participants were studied at D15S128, which lies within the SNRPN IC region.
PCR amplification and sequencing analysis
The ExonPrimer program was used to design PCR primers flanking each exon of the 11 candidate genes. Sequences were obtained from the chromosome 15q11q13 draft human genome sequence (primer sequences are available on request). Direct sequencing was performed using the dideoxy chain termination method (Applied Biosciences, ABI BigDye 3.0 system) on an ABI 3730 DNA Sequencer, and sequences were analysed using Chromas v2.0 software. All base variations were verified bidirectionally.
Expression analysis
Prior to the isolation of cDNA, the RNA samples were treated with DNAse1 to ensure that they did not contain any contaminating genomic DNA. cDNA was then isolated by standard methods using a Taqman reverse transcription kit (Applied Biosciences, Warrington, UK). Expression of candidate genes was then measured by quantitative methodology using SYBR green as the detector. As SYBR green will only fluoresce when it is bound to double-stranded DNA, the point at which the concentration of PCR product is above a set background threshold will be related to the amount of cDNA template in the PCR mix. The actual product concentration is estimated by constructing a standard curve produced by serial dilutions of cDNA from a control individual. To allow for differences in cDNA concentration, these values are normalised against those from a housekeeping gene such as β-actin (BA) or GAPDH. Thus the level of expression of any sample can be calculated for different types of proband, relative to controls. Expression analysis was performed using an Applied Biosystems ABI prism 7900HT sequence detection system. Primers were derived using the programme Primer 3. Where possible, primer pairs crossing an intron were selected. Those used for SNRPN crossed intron 6.
Results
A total of nine participants from the Cambridge, UK, PWS studies (Whittington et al. 2001; Soni 2006) were recruited into the subset. These participants all had a confirmed clinical and behavioural diagnosis of PWS but presented with unusual genetics.
Clinical description and genetic studies
The clinical details for all participants are shown in Table 1. All were examined by at least two of the authors and were found to fulfil the criteria for a clinical diagnosis of PWS as described by Holm et al. (1993).
Participant id015
This person (case 1 in Whittington et al. 2002) demonstrated both a maternally and a paternally derived band at the SNURF/SNRPN exon 1 region by methylation-specific PCR (MS-PCR) and also expressed the SNRPN gene when studied by reverse transcriptase (RT-PCR) (Fig. 1; Table 2). However, a series of microsatellite markers across the 15q11q13 region revealed a small paternally derived deletion located proximal to SNRPN. It included D15S543, D15S18 and D15S9 (MKRN3) but neither D15S541 nor D15S11. Thus the maximum size of the deletion was 1.8 Mb (Fig. 2). Microsatellite analysis at a series of loci extending throughout the remainder of the PWS/AS region demonstrated both maternal and paternal alleles (Tables 3, 4), although he was uninformative at D15S128. Expression studies carried out on a series of candidate genes showed that this proband expressed HBII-85 and PAR5 at levels comparable to those shown by control subjects (Table 2), while sequencing studies showed SNPs close to exons 2, 7 (rs705) and 12 of the SNRPN gene but no inactivating mutation in any candidate gene located in the PWS region (Table 5).

Methylation-specific PCR of the SNURF/SNRPN exon 1 region. Panel ALane 1 100-bp ladder, lane 2 id119. Panel BLane 1 1-kb ladder, lane 2 id 015. Maternally derived methylated band is at 313 bp, paternally derived non-methylated band is at 221 bp, both are present in id119 and id015

Microsatellite map of the nine probands showing the size and the relative positions of the deletions. These are maximum deletions in that if the locus is mono-allelic it has been included even if it is uninformative. Distances are given in megabases from the centromere, and the hatched region defines the limit of the deletion
Participant id027
This person also presented with classical PWS. She fulfilled the genetic diagnostic criteria as she did not express SNRPN and had only a maternally derived non-methylated band after MS-PCR at SNURF/SNRPN exon 1. A paternal sample was unavailable, but while the proband was mono-allelic across a region that included D15S11, D15S817, and D15S1021, she carried a non-maternally derived allele at other loci including D15S128, D15S1513 and GABRB3 that lie within the PWS region (Tables 3, 4). These data suggest that either id027 has a smaller than usual deletion in 15q11q13 (Fig. 2) or the proband has an IC epimutation. She carried variants near SNRPN exon 12 and IPW exon 3 (Table 5).
Participants id026, id090, id094 and id096
These four people fulfilled the criteria for a clinical diagnosis of PWS, but each was found to carry an IC mutation. All four have only the maternal methylated band at SNURF/SNRPN exon 1, and none of them expresses SNRPN. Parental samples were obtained in each case, and microsatellite analysis showed bi-allelic inheritance throughout most of the 15q11q13 region (Tables 3, 4). Participants id026 (case 4 in Whittington et al. 2002) and id096 had only a maternally inherited copy of D15S128, but id090 and id094, although mono-allelic, were uninformative at this locus.
All four demonstrated a paternal allele at informative loci throughout the region (Tables 3, 4). The maximum extent of the deletion carried by each of them is shown in Fig. 2. Subject id026 had only the C allele at rs220030 lying within the SNURF/SNRPN minimal promoter region and participants id090, id094 and id096 only the T allele.
None of these four people had an inactivating mutation in any of the candidate genes studied (Table 5).
Participant id81
This person entered the study during its second phase (Soni 2006). Both a paternally derived and a maternally derived band could be detected after MS-PCR at SNURF/SNRPN exon 1. Parental samples were not available, but this person was also bi-allelic at microsatellite loci throughout the PWS/AS region including D15S128 (Tables 3, 4). Quantitative RT-PCR detected expression of SNRPN at a reduced level (Table 2). Expression of UBE3A and ATP10C, which are located within the AS rather than the PWS region, were within the normal range indicating that the reduced expression levels were confined to the PWS part of the region. He was heterozygous C/T at SNP rs220030, which is the only one recorded as located within the SNURF/SNRPN minimal promoter region (Table 5).
Participant id119
This participant is a member of a family in which an intrachromosomal inversion of 15p11q12 segregates (Webb et al. 1992; Clayton-Smith et al. 1993), and he has a cousin with AS. Microsatellite analysis of id119 and his parents showed that he carried a paternal deletion at several loci in the PWS/AS region including D15S128, and he did not express SNRPN (Tables 2, 3, 4). However repeated MS-PCR of SNURF/SNRPN exon 1 always revealed the presence of both a methylated and a non-methylated band (Fig. 1), and sequencing of the SNRPN minimal promoter showed that id119 had only the T allele at rs220030 and a G → A transition at base 22,751,225 which is the fourth base prior to the start of transcription of the SNURF/SNRPN gene (Fig. 3). This change was confirmed by bi-directional sequencing. A paternal sample showed only the normal G allele, but the maternal sample showed that she was a heterozygous carrier G/A (Fig. 3).

Sequencing of part of the SNURF/SNRPN minimal promoter region in the family of id119. The change is indicated by arrows. a id119 forward. b id119 reverse. c Father of id119 forward. d Mother of id119 forward
Participant id153
Despite fulfilling the clinical criteria for a diagnosis of PWS (case 2 in Whittington et al. 2002), MS-PCR at SNURF/SNRPN exon 1 demonstrated both a paternally derived non-methylated band and a maternally derived methylated band, precluding a laboratory diagnosis of PWS. Although parental samples were not available, this person also demonstrated bi-allelic inheritance throughout the entire PWS/AS region including D15S128 (Tables 3, 4). Sequencing detected SNPs near to the SNRPN gene exons 7 (rs705) and 12, but no inactivating mutations were found in any candidate gene, including the SNURF/SNRPN minimal promoter. Despite this, quantitative RT-PCR demonstrated that, although expression of SNRPN was detected, it was at a reduced level. Low expression was also found at certain other loci within the PWS region including PAR5 and HBII-85 (Table 2).
Expression analysis
None of the people who demonstrated a single maternally derived methylated band at the SNRPN locus expressed SNRPN, but the person with a very small deletion, id015, did so with a value within the normal range. The two probands who had a firm clinical, but not a genetic, diagnosis of PWS also expressed this transcript but at a reduced level. The average expression level for control participants was 1.1 ± 0.4, whereas for id153, the average value obtained over five separate measurements, each performed in triplicate, was 0.4, and for id81 it was 0.5 over four separate measurements, each performed in triplicate. These values were significantly different from those obtained for a series of unaffected control participants when subjected to the student’s t test. Also the average control value for the relative expression of HBII-85 was 1.5 ± 1.4, for id153 it was 0.7, and for id81 it was 0.5. The wider range of values obtained for HBII-85 reflects the difficulties in detecting expression quantitatively in the more distal part of the PWS region. In the AS region, however, neither participant showed reduced values for UBE3A or ATP10C (Table 2).
Discussion
Mutation analysis has been carried out on 11 imprinted genes located within 15q11q13 using DNA from nine participants who clinically have PWS but who do not fulfil the genetic diagnostic criteria. If the proband is >3 years of age, a clinical diagnosis of PWS will include a score of 8 points, of which 4 should be from the eight major diagnostic criteria (Holm et al. 1993; Cassidy 1997). Two of the authors, who examined all of the participants who entered the Cambridge study, were convinced that id81 and id153 were clinically indistinguishable from those with a confirmed laboratory diagnosis of PWS (both were >40 years of age). Mosaicism was not detected. They not only expressed SNRPN and HBII-85 (Ding et al. 2005), although possibly at a reduced level, as shown by quantitative real-time PCR, but were also heterozygous for loci within the deletion region. While the latter does not eliminate an imprinting defect, expression of SNRPN usually does. These results suggested that the signs and symptoms of PWS in these participants could be caused by another genetic abnormality, such as a mutation in the SNRPN gene, particularly in the minimal promoter region (SMP/exon1) containing the putative imprinting control centre, or possibly in any other paternally expressed gene including SNURF, HBII-85, HBII-13, HBII-436, HBII-438a, IPW, PAR1, PAR5, NECDIN and MKRN3.
As no DNA mutation was found in any of these people, and naturally occurring SNP polymorphisms do not seem to be associated with clinical symptoms (Oeffner et al. 2001), their PWS phenotype could be attributed to a phenocopy, a gene located elsewhere in the genome (D’Angelo et al. 2006), or to partial expression of the epimutation error described by El-Maarri et al. (2001) in people with IC mutations but without a microdeletion, or it may be that both probands lie at the low expressing end of a normal distribution. If as El-Maarri et al. (2001) have suggested, the majority of people with an IC fail to reset the parental imprint correctly after fertilisation rather than in the gamete, or in other cases fail to erase the maternal imprint during spermatogenesis (Buiting et al. 2003), either explanation could account for the reduced expression of SNRPN in these two people.
The loss in id015 of a small upstream, paternally derived part of the PWSCR with apparently no involvement of the IC, but accompanied by clinical manifestation of PWS, would suggest that despite the genetic evidence, any disruption of the imprinted region may result in the syndrome. Even a maternal deletion encompassing only the ATP10C gene has been associated with PWS-like symptoms (Ninomiya et al. 2005). In contrast, id015 was found to over-express both UBE3A and ATP10C, the two paternally imprinted genes associated with AS. As this tendency was also detected in some other participants with unusual genetics (Table 2) and UBE3A is associated with a paternally expressed antisense transcript, it is tempting to suggest that the over-expression is related to the PWS phenotype.
In one proband (id119), sequencing of the SMP/exon1 region of the SNRPN gene revealed a point mutation (G → A) at position −4 from the start of transcription of SNRPN. He also has a paternally inherited deletion that extends throughout the entire PWS/AS region, providing an explanation for his clinical diagnosis of PWS. As he has only the A allele, his father only the G and his mother is heterozygous A/G, then the change in sequence must be maternally inherited. In addition, id119 also consistently demonstrated a non-methylated band at the SNRPN promoter region when investigated by MS-PCR and was considered clinically to be relatively mildly affected by PWS. These three factors are likely to be linked. The mutation is not in one of the important 7-bp elements (SBE) described by Ohta et al. (1999) but is closer to the start of transcription than any of them. It is likely not only that the non-methylated band is maternally derived but also that the G → A mutation is responsible for a failure to reset the imprinting pattern completely. Such “leaky” imprinting could also account for the milder than expected clinical picture presented by id119.
Although there are many differences between the mouse and human SNRPN genes, the striking similarities validate the use of mouse models. In the mouse, the methylation pattern of the maternally inherited homologue is erased at an early stage of gametogenesis and established either during oogenesis (Shemer et al. 1997) or, as has been shown in cases of IC deletion, after fertilisation (El-Maari et al. 2001).
Ohta et al. (1999), while studying three patients with an imprinting mutation but with no detectable mutation, identified the SMP/exon1 region as the PWS-IC. However Shemer et al. (2000), using mouse models, found that the SMP/exon1 alone was insufficient but needed the cis-acting AS-SRO both for the removal of the maternal methylation imprint and for promoter expression. By using a transgene consisting of human AS-SRO and mouse SMP/exon1, they were able to establish that the maternal allele was methylated and not expressed while the paternal allele was unmethylated and expressed. A series of papers then dissected the transgene using the CAT expression vector to determine expression and methylation-sensitive enzymes to determine the extent of methylation.
Ohta et al. (1999), Hershko et al. (2001), and Finberg et al. (2003), using AS-SRO-mouse SMP mini-transgenes to establish and maintain the imprinted state, found an SBE (7-bp element) within the SMP to be an absolute requirement for promoter activity. Differences between mouse and human imprinting mechanisms were revealed when the orthologous human SMP, which has sequence-identical but not positional-identical SBEs, failed to methylate the maternal allele. The mouse SBE, when unmethylated, bound a specific protein required for promoter activity. Mutation of the SBE in the mouse did not completely abolish methylation of the maternal allele indicating that sequences outside the SBE are also required. AS-SRO-human-SMP/exon1 did not induce methylation of the promoter upon maternal transmission, and AS-SRO-human SMP/mouse exon1 had neither promoter activity nor methylated state on female transmission.
The same group (Kantor et al. 2004), using five different mini-transgenes, isolated five cis elements involved in different steps of the imprinting process. The original SMP consisted of −84 bp upstream/exon1. If base pairs −84 to −42 were removed then the maternal allele did not become methylated during oogenesis. If bp +1 to +40 were removed again, loss of maternal methylation resulted. So de novo methylation signals map both to upstream sequences of the SMP and to exon1 of the gene itself, and both are required.
Buiting et al. (2003) studied 51 patients with PWS and an IC mutation. Sequencing of the SMP/exon1 in 32 patients without an IC deletion revealed no point mutations, but 7/32 had a C → G change at bp−83. All were heterozygous, and the change was found in 7/68 controls. The G → A change at bp−4 was also detected in one PWS patient. Because it was also found in the mother and in a single proband with AS without a deletion (although not in a series of controls), it was considered to be a neutral variant. However, this patient differed from id119 in that he/she did not have a paternally transmitted deletion, which accounted for the clinical picture of PWS, but all paternal sequences were apparently intact.
The integrity of the SMP/exon1 region is vital for establishing and maintaining the imprinted state in both mice and humans, and upstream sequences are critical for the correct function of either mechanism. Changes in the non-coding regions can cause genetic disease by altering gene expression (Buckland 2004). We suggest that in the case of id119, the change from G → A at bp-4 alters the binding of a factor, such that methylation of the maternal allele is not fully established, and results in a non-methylated band during MS-PCR, together with partial expression of SMP/exon1. This may explain the relatively mild clinical picture presented by this person.
A similar situation has recently been described by Wu et al. (2006). They showed that mutations upstream of Snrpn-exon1 caused lack of methylation in the maternal Snrpn promoter and activation of maternally expressed genes including a rescue from the lethality and growth retardation normally shown by the PWS mouse.
DNA sequence variants such as the one described here would affect the binding of factors that are involved in establishing the correct epigenetic state (Lee and Wevrick 2000). Genetic factors have been found to be responsible for an increased risk of an imprinting defect in AS (Buiting et al. 2001) and are considered to be responsible for the majority of individual, parentally dependent variability in epigenetic modification in other regions of the genome (Sandovici et al. 2003). In a recent study of childhood absence epilepsy, Urak et al. (2006) were able to demonstrate that certain promoter haplotypes present in the GABRB3 gene were not only associated more strongly with the disease but at the same time lowered the transcriptional activity of the putative disease gene.
In summary, a study of nine probands who clinically had PWS but with atypical genetics has suggested that changes in the maternal SMP may alleviate some of the more severe manifestations of the disease, that PWS can result from a stochastic partial change in gene expression, and that even minor disruption of the PWSCR outside the SRO can still cause PWS.
References
Amos-Landgraf JM, Yonggang J, Gottlieb W, Depinet T, Wandstrat AE, Cassidy SB, Driscoll DJ, Rogan PK, Schwartz S, Nicholls RD (1999) Chromosome breakage in the Prader-Willi and Angelman syndromes involves recombination between large, transcribed repeats at proximal and distal breakpoints. Am J Hum Genet 65:370–386
Boccaccio I, Glatt-Deeley H, Watrin F, Roeckel N, Lalande M, Muscatelli F (1999) The human MAGEL2 gene and its mouse homologue are paternally expressed and mapped to the Prader-Willi region. Hum Mol Genet 8:2497–2505
Buckland PR (2004) Allele specific gene expression differences in humans. Hum Mol Genet 13:255–260
Buiting K, Barnicoat A, Lich C, Pembrey M, Malcolm S, Horsthemke B (2001) Disruption of the bipartite imprinting centre in a family with Angelman syndrome. Am J Hum Genet 68:1290–1294
Buiting K, Gross S, Lich C, Gillessen-Kaesbach G, El-Maarri O, Horsthemke B (2003) Epimutations in Prader-Willi and Angelman syndromes: a molecular study of 136 patients with an imprinting defect. Am J Hum Genet 72:571–577
Cassidy SB (1997) Prader-Willi syndrome. J Med Genet 34:917–923
Cavaille J, Buiting K, Kiefmann M, Lalande M, Brannan CI, Horsthemke B, Bachellerie J-P, Brosius J, Huttenhofer A (2000) Identification of brain-specific and imprinted small nucleolar RNA genes exhibiting an unusual genomic organisation. Proc Natl Acad Sci USA 97:14311–14316
Chai JH, Locke DP, Greally JM, Knoll JHM, Ohta T, Dunai J, Yavor A, Eichler EE, Nicholls RD (2003) Identification of four highly conserved genes between breakpoint hotspots BP1 and BP2 of the Prader-Willi/Angelman syndromes deletion region that have undergone evolutionary transposition mediated by flanking duplicons. Am J Hum Genet 73:898–925
Christian SL, Robinson WP, Huang B, Mutirangura A, Line MR, Nakao M, Surti V, Chakravarti A, Ledbetter DH (1995) Molecular characterization of two proximal deletion breakpoint regions in both Prader-Willi and Angelman syndrome patients. Am J Hum Genet 57:40–48
Clayton-Smith J, Driscoll DJ, Waters MF, Webb T, Andrews T, Malcolm S, Pembrey ME, Nicholls RD (1993) Difference in methylation patterns within the D15S9 region of chromosome 15q11–13 in first cousins with Angelman syndrome and Prader-Willi syndrome. Am J Med Genet 47:683–686
D’Angelo CS, Da Paz JA, Kim CA, Bertola DR, Castro CIE, Varela MC, Koiffmann CP (2006) Prader-Willi-like phenotype: investigation of 1p36 deletion in 41 patients with delayed psychomotor development, hypotonia, obesity and/or hyperphagia, learning disabilities and behavioural problems. Eur J Med Genet 49(6)451–460
de los Santos T, Schweizer J, Rees CA, Francke U (2000) Small evolutionarily conserved RNA, resembling C/D box small nucleolar RNA, is transcribed from PWCR1, a novel imprinted gene in the Prader-Willi deletion region which is highly expressed in brain. Am J Hum Genet 67:1067–1082
Ding F, Prints Y, Dhar MS, Johnson DK, Garnacho-Montero C, Nicholls RD, Francke U (2005) Lack of Pwcr1/MBII-85 snoRNA is critical for neonatal lethality in Prader-Willi syndrome mouse models. Mamm Genome 16:424–431
El-Maarri O, Buiting K, Peery EG, Kroisel PM, Balaban B, Wagner K, Urman B, Heyd J, Lich C, Brannan CI, Walter J, Horsthemke B (2001) Maternal methylation imprints on human chromosome 15 are established during or after fertilisation. Nature Genet 27:341–344
Finberg YG, Kantor B, Hershko AY, Razin A (2003) Characterisation of the human SNRPN minimal promoter and cis elements within it. Gene 304:201–206
Gallagher RC, Pils B, Albalwi M, Francke U (2002) Evidence for the role of PWCR1/HBII-85 C/D box small nucleolar RNAs in Prader-Willi syndrome. Am J Hum Genet 71:669–678
Glenn CC, Saitoh S, Jong MT, Filbrandt MM, Surti U, Driscoll DJ, Nicholls RD (1996) Gene structure, DNA methylation and imprinted expression of the human SNRPN gene. Am J Hum Genet 58:335–346
Gray TA, Saitoh S, Nicholls RD (1999) An imprinted mammalian bicistronic transcript encodes two independent proteins. Proc Natl Acad Sci USA 96:5616–5621
Hershko AY, Finberg Y, Kantor B, Shemer R, Razin A (2001) The mouse SNRPN minimal promoter and its human orthologue: activity and imprinting. Genes Cells 6:967–975
Holland AJ, Treasure J, Coskeran P, Dallas J (1995) Characterisation of the eating disorder in the Prader-Willi syndrome. J Intellect Dis Res 39:377–378
Holland A, Whittington J, Hinton E (2003) The paradox of Prader-Willi syndrome: a genetic model of starvation. Lancet 362:989–991
Holm VA, Cassidy SB, Butler MG, Hanchett JM, Greenswag LR, Whitman BY, Greenberg F (1993) Prader-Willi syndrome: consensus diagnostic criteria. Pediatrics 91:398–402
Jay P, Rougeulle C, Massacrier A, Moncla A, Mattei M-G, Malzac P, Roeckel N, Taviaux S, Lefranc JL, Cau P, Berta P, Lalande M, Muscatelli F (1997) The human necdin gene, NDN, is maternally imprinted and located in the Prader-Willi syndrome chromosomal region. Nat Genet 17:357–361
Jong MTC, Gray TA, Yonggang J, Glenn GC, Saitoh S, Driscoll DJ, Nicholls RD (1999) A novel imprinted gene, encoding a RING zinc-finger protein, and overlapping antisense transcript in the Prader-Willi syndrome critical region. Hum Mol Genet 8:783–793
Kantor B, Makedonski K, Green-Finberg Y, Shemer R, Razin A (2004) Control elements within the PWS/AS imprinting box and their function in the imprinting process. Hum Mol Genet 13:751–762
Kishino T, Lalande M, Wagstaff J (1997) UBE3A/E6-AP mutations cause Angelman syndrome. Nat Genet 15:70–73
Kubota T, Das S, Christian SL, Baylin SB, Herman JG, Ledbetter DH (1997) Methylation-specific PCR simplifies imprinting analysis. Nat Genet 16:16–17
Landers M, Bancescu DL, Le Meur E, Rougeulle C, Glatt-Deeley H, Brannan C, Muscatelli F, Lalande M (2004) Regulation of the large (approximately 1,000 kb) imprinted murine Ube3a antisense transcript by alternative exons upstream of SNURF/SNRPN. Nucleic Acids Res 32:3480–3492
Lee S, Wevrick R (2000) Identification of novel imprinted transcripts in the Prader-Willi syndrome and Angelman syndrome deletion region: further evidence for regional imprinting control. Am J Hum Genet 66:848–858
Lee S, Kozlov S, Hernandez L, Chamberlain SJ, Brannan CI, Stewart CL, Wevrick R (2000) Expression and imprinting of MAGEL2 suggest a role in Prader-Willi syndrome and the homologous murine imprinting phenotype. Hum Mol Genet 9:1813–1819
MacDonald HR, Wevrick R (1997) The necdin gene is deleted in Prader-Willi syndrome and is imprinted in human and mouse. Hum Mol Genet 6:1873–1878
Nicholls RD, Knepper JL (2001) Genome organisation, function and imprinting in Prader-Willi and Angelman syndromes. Annu Rev Genomics Hum Genet 2:153–175
Ninomiya S, Yokoyama Y, Kawakami M, Une T, Maruyama H, Morishima T (2005) Unique maternal deletion of 15q in a patient with some symptoms of Prader-Willi syndrome. Pediatr Int 47:541–545
Oeffner F, Korn T, Roth H, Ziegler A, Hinney A, Goldschmidt H, Siegfried W, Hebebrand J, Grzeschik K-H (2001) Systematic screening for mutations in the human necdin gene (NDN): identification of two naturally occurring polymorphisms and association analysis in body weight regulation. Int J Obes 25:767–769
Ohta T, Gray TA, Rogan PK, Buiting K, Gabriel JM, Saitoh S, Muralidhar B, Bilienska B, Krajewska-Walasek M, Driscoll DJ, Horsthemke B, Butler MG, Nicholls RD (1999) Imprinting-mutation mechanisms in Prader-Willi syndrome. Am J Hum Genet 64:397–413
Pagliardini S, Ren J, Wevrick R, Greer J (2005) Developmental abnormalities of neuronal structure and function in prenatal mice lacking the Prader-Willi syndrome gene necdin. Am J Pathol 167:175–191
Rougeulle C, Glatt H, Lalande M (1997) The Angelman syndrome candidate gene UBE3A/E6-AP is imprinted in brain. Nature Genet 17:14–15
Rougeulle C, Cardoso C, Fontes M, Colleaux L, Lalande M (1998) An imprinted antisense RNA overlaps UBE3A and a second maternally expressed transcript. Nature Genet 19:15–16
Runte M, Huttenhofer A, Gross S, Kiefmann M, Horsthemke B, Buiting K (2001) The IC-SNURF-SNRPN transcript serves as a host for multiple small nucleolar RNA species and as an antisense RNA for UBE3A. Hum Mol Genet 10:2687–2700
Sandovici I, Leppert M, Hawk PR, Suarez A, Linares Y, Sapienza C (2003) Familial aggregation of abnormal methylation of parental alleles at the IGF2/H19 and IGF2R differentially methylated regions. Hum Mol Genet 12:1569–1578
Shemer R, Birger Y, Riggs AD, Razin A (1997) Structure of the imprinted mouse Snrpn gene and establishment of its parental-specific methylation pattern. Proc Natl Acad Sci USA 94:10267–10272
Shemer R, Hershko AY, Perk J, Mostoslavsky R, Tsuberi B, Cedar H, Buiting K, Razin A (2000) The imprinting box of the Prader-Willi/Angelman Syndrome domain. Nat Genet 26:440–443
Soni S (2006) An investigation into psychiatric illness in people with Prader-Willi syndrome: evidence for a genetic basis for psychosis. PhD Thesis, Cambridge University, Cambridge
Urak L, Feucht M, Fathi N, Hornik K, Fuchs K 2006) A GABRB3 promoter haplotype associated with childhood absence epilepsy impairs transcriptional activity. Hum Mol Genet 15:2533–2541
Webb T, Clayton-Smith J, Cheng X-J, Knoll JHM, Lalande M, Pembrey ME, Malcolm S (1992) Angelman syndrome with a chromosomal inversion 15inv(p11q13) accompanied by a deletion in 15q11q13. J Med Genet 29:921–924
Wevrick R, Francke U (1996) Diagnostic test for the Prader-Willi syndrome by SNRPN expression in blood. Lancet 348:1068–1069
Whittington JE, Holland AJ, Webb T, Butler J, Clarke D, Boer H (2001) Population prevalence and estimated birth incidence and mortality rate for people with Prader-Willi syndrome in one UK health region. J Med Genet 38:792–798
Whittington JE, Holland AJ, Webb T, Butler J, Clarke D, Boer H (2002) Relationship between clinical and genetic diagnosis of Prader-Willi syndrome. J Med Genet 39:926–932
Wu M-Y, Tsai T-F, Beaudet AL (2006) Deficiency of Rbbp1/Arid4a and Rbbp1l1/Arid4b alters epigenetic modifications and suppresses an imprinting defect in the PWS/AS domain. Genes Develop 20:2859–2870
Zeschnigk M, Lich C, Buiting K, Doerfler W, Horsthemke B (1997) A single-tube PCR test for the diagnosis of Angelman and Prader-Willi syndrome based on allelic methylation differences at the SNRPN locus. Eur J Hum Genet 5:94–98
Acknowledgements
This work was supported by the Wellcome Trust, the Health Foundation and the UK PWS association. We are extremely grateful to all of the people with PWS, their families and their caregivers who participated in this study. Ethical approval for collection and analysis of blood samples for genetic studies on PWS was obtained from Eastern MREC.
Author information
Authors and Affiliations
Corresponding author
URLS and genome browsers used in this study
URLS and genome browsers used in this study
Rights and permissions
About this article
Cite this article
Maina, E.N., Webb, T., Soni, S. et al. Analysis of candidate imprinted genes in PWS subjects with atypical genetics: a possible inactivating mutation in the SNURF/SNRPN minimal promoter. J Hum Genet 52, 297–307 (2007). https://doi.org/10.1007/s10038-007-0109-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-007-0109-6
Keywords
This article is cited by
-
Behavioral profile of adults with Prader-Willi syndrome: correlations with individual and environmental variables
Journal of Neurodevelopmental Disorders (2013)
-
Different distribution of the genetic subtypes of the Prader–Willi syndrome in the elderly
European Journal of Human Genetics (2010)
