
Abstract
In this study, we take advantage of a natural experiment—a 2004 mass die-off of the Common Murre in Alaska to determine whether closely related mtDNA haplotypes differ in their probability of being eliminated during such a short term but a marked event removing hundreds of thousands of individuals. We sequenced complete mtDNA ND2 gene (1041 bp) for 168 Common Murres sampled from seven breeding colonies across Alaska before the 2004 die-off and 127 dead murres washed ashore during the die-off. We found little mtDNA variation and lack of geographical structuring among the seven Common Murre breeding colonies in Alaska. A comparison of the single-dominant mtDNA haplotype's frequency between live murres sampled on breeding colonies before the die-off (73.2%; 95% confidence interval 66.3–79.9%) and dead murres sampled during the die-off (59.1%; 95% confidence interval 50.4–67.4%; Fisher's exact P=0.01) showed that carriers of the dominant haplotype were significantly less likely to die than carriers of other haplotypes. At the same time, the ratio of non-synonymous to synonymous substitutions did not differ between live (10:35) and dead birds (18:34; Fisher's exact P=0.26), indicating that non-synonymous substitutions were as likely to be eliminated as synonymous substitutions. These results are consistent with the possibility of positive selection on the dominant mtDNA haplotype during the die-off.
Similar content being viewed by others
Introduction
Many vertebrates exhibit marked population cycles when periods of population growth end, with population crashes eliminating a large number of individuals. The Common Murre Uria aalge is an example of such a species. It is an abundant seabird of the Northern Hemisphere but occasionally it experiences so-called ‘seabird wrecks’ when tens of thousands of starved murres are washed ashore (Ainley et al., 2002). Although these die-offs happen throughout the species' range, their amplitude in Alaska is very large and includes hundreds of thousands of individuals (Gromme, 1927; Bailey and Davenport, 1972; Piatt and van Pelt, 1997). These mass die-offs occur in winter to early spring in the Gulf of Alaska where most of the Alaskan murres spend winter. During a single die-off in 1993, an estimated minimum of 250 000 Common Murres washed ashore within a few months. This is likely a considerable underestimate of the total mortality because many more birds could have been carried to the open ocean or sunk before reaching the shore (Piatt and van Pelt, 1997). The minimum estimate of 250 000 individuals corresponds to ca. 9% of the total population of Alaskan Common Murres, which is estimated at 2.8 × 106 (U.S. Fish and Wildlife Service, 2006a). Mass die-offs of the Common Murre appear to result from periodic collapses of schooling prey (Vader et al., 1990). The frequency of these die-offs, the last two die-offs occurred 11 years apart in 1993 and 2004, appears to exceed the generation time of the Common Murre, which start breeding between 2 and 7 years of age (Ainley et al., 2002).
A recent multilocus genetic study of the Common Murre (Morris-Pocock et al., 2008) showed a lack of geographical structuring in the mtDNA control region, nuclear intron sequences and microsatellite frequencies across the entire Pacific range of the species. The nucleotide diversity of the Common Murre mtDNA control region sequences from the Pacific was half that of the Atlantic regardless of whether colonies were considered individually or combined into ocean basins. Furthermore, comparative multilocus genetic study of three species of alcids (Alcidae) in the Pacific (Friesen and Piatt, 2003) showed that the Common Murre, Pigeon Guillemot Cepphus columba and Marbled Murrelet Brachyramphus marmoratus had similar levels of nuclear intron sequence and microsatellite variation, whereas mtDNA control region was three times less variable in the Common Murre than in the other two seabirds. The lack of geographical structuring combined with low mtDNA variation in the Pacific compared with Atlantic Common Murres, similar levels of the nuclear DNA variation but several fold lower mtDNA variation in Pacific Common Murres than in other alcids, suggest that the periodic die-offs could be responsible for the low mtDNA variation in the Pacific Common Murre. Demographic declines are expected to have a greater effect on the mtDNA variation than on the nuclear DNA variation because of the fourfold lower effective population size of mtDNA. The compelling question, however, is whether mtDNA haplotypes are eliminated at random or do they differ in their probability of surviving a catastrophic event such as a mass die-off.
To test this question, we compared mtDNA ND2 gene (1041 bp) haplotype frequencies between live Common Murres sampled on seven breeding colonies across Alaska and dead Common Murres washed ashore during the last mass die-off. If the mtDNA haplotypes are eliminated at random, we would not expect significant differences in haplotype frequencies or in the non-synonymous to synonymous substitutions ratio between the live and dead murres. The lack of significant difference in the frequency of the common haplotype(s) but a significant difference in the non-synonymous to synonymous substitutions ratio would be consistent with purifying selection, which is expected to preferentially eliminate non-synonymous substitutions. A significant underrepresentation of a single common haplotype among the dead birds and similar non-synonymous to synonymous substitutions ratio among the rare haplotypes in live and dead birds would be consistent with a selective advantage of the most common haplotype.
Materials and methods
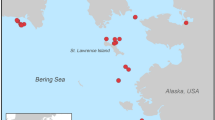
Although the migration of Common Murres is not well-known, birds breeding in the Bering Sea and Gulf of Alaska winter in the northern Gulf of Alaska where they form very large ‘floats’ (Ainley et al., 2002). This wintering area was affected by the 2004 die-off. During this last die-off of Common Murres, on April 20 2004, SVD collected wing muscle tissue from 127 dead murres near Whittier, Alaska. During 2000, 2002 and 2003 breeding seasons, ASK collected blood samples from 168 Common Murres and 23 Thick-billed Murres Uria lomvia captured live on seven different breeding colonies in Alaska (Figure 1). The Thick-billed Murre is the sister species of the Common Murre. It is almost equally abundant in Alaska but is not affected by mass die-offs (Vader et al., 1990) so it is the best species available as an outgroup and control for comparison of mtDNA diversity.

Sampling years, localities and sizes for Common (cm), and Thick-billed Murres (tbm). Pie charts represent proportions of the dominant (white) and mutant haplotypes (gray, synonymous mutations only; black, at least one non-synonymous substitution) for dead (a) and live (b) Common Murres.
For all individuals we sequenced the complete mtDNA ND2 gene (1041 bp). Total genomic DNA was extracted using the DNeasy Blood and Tissue Kit (QIAGEN, Valencia, CA, USA). We used GoTaq Green Master Mix (Promega, Madison, WI, USA) and primers L5215: 5′-TATCGGGCCCATACCCCGAAAAT-3′ (Hackett, 1996) and H1064: 5′-CTTTGAAGGCCTTCGGTTTA-3′ (Drovetski et al., 2004) for fragment amplification via PCR. The PCR profile included 2.5 min preheating at 94 °C and 35 cycles of 30 s at 94 °C, 30 s at 57 °C and 45 s at 72 °C. PCR cycles were followed by 10 min of extension at 72 °C. The PCR fragments were sequenced directly on ABI-3100 or ABI-3730 sequencers using BigDye chemistry (Applied Biosystems, Foster City, CA, USA) and amplification primers.
Sequences were aligned automatically using Sequencher 4.9 (Gene Codes Corporation, Ann Arbor, MI, USA). The alignment did not require editing because there were no indels in the ND2 sequences. NCBI accession numbers for the Common Murre sequences are JQ434767–JQ435061, and for the Thick-billed Murre are JQ435062–JQ435084.
Unique haplotypes were identified using DnaSP 5.10 (Librado and Rozas, 2009). The proportion of dominant haplotypes was calculated for live and dead birds separately and 95% confidence intervals (CIs) were calculated for each proportion. Bootstrapping was used to construct the CIs, by resampling with replacement, 10 000 times (Efron and Tibshirani, 1986).
We statistically tested the equivalence of dominant haplotype frequencies between live and dead birds using Fisher's exact test implemented in StatView 5.0 (SAS Institute Inc., Cary, NC, USA), a non-parametric bootstrapping approach (Efron and Tibshirani, 1993) and a traditional two-sample test of proportions, which uses a normal approximation to the binomial distribution (Zar, 1996). Bootstrapping was used to non-parametrically determine the expected range of values under the null hypothesis, live and dead birds having the same dominant haplotype frequency, to determine statistical significance at the 0.05 level. We then compared our observed dominant haplotype frequency for the dead birds to the 95% CI based on the null hypothesis. Using the sample of 168 live birds as a representation of the total population, we simulated the selection of 127 birds (our sample of dead birds) 10 000 times, resampling with replacement. The resampling was performed using SAS 9.1 (SAS Institute Inc.) with a random seed. The frequency of the dominant haplotype was calculated for each sample, and the 95% CI was estimated as the 2.5 and 97.5th percentile of the resulting values.
We used DnaSP for calculating haplotype (Hd) and nucleotide (πn) diversity and their s.d., and for conducting intraspecific neutrality/demographic history tests: Tajima (1989, 1996), Fu's Fs (Fu, 1997), R2 (Ramos-Onsins and Rozas, 2002) and interspecific neutrality McDonald–Kreitman (MK) test (McDonald and Kreitman, 1991). These tests elucidate long-term demographic/evolutionary processes affecting murre populations over many thousands of generations. Understanding the general evolutionary background is important for identification of potential biases in our haplotype frequency analyses. We used Arlequin 3.11 (Excoffier et al., 2005) to conduct an analysis of molecular variance (Excoffier et al., 1992), to compute pairwise Fst values among localities, and perform a Mantel's test (Mantel, 1967) for isolation by distance among breeding colonies. A maximum likelihood phylogenetic analysis was performed using PAUP* 4.0 (Swofford, 1998). The maximum likelihood models and parameters were determined using Akaike information criterion (Akaike, 1974) implemented in jModeltest 3.7 (Posada, 2008).
Results
DnaSP identified 9 haplotypes among 23 Thick-billed Murres and 54 haplotypes among 295 Common Murres. Our maximum likelihood analysis resulted in a single tree (Figure 2) with two clades. One clade consisted of all Common Murre haplotypes, the other of all Thick-billed Murre haplotypes. The distance between these clades was 6.95%. The shape of both murre clades was star-like, with the most common haplotype at the center and majority of rare haplotypes only a single-mutation step away.

Maximum likelihood tree of Common and Thick-billed Murre haplotypes. White circles represent live individuals sampled at the breeding colonies before the mass die-off, black circles represent individuals that were washed ashore during the die-off. The area of the circles represents the frequency of haplotypes.
Pairwise Fst values for all possible locality pairs varied from −0.037 to 0.079 (Table 1). Their P-values were >0.05 for all locality pairs except for Bogoslof and Gull islands for which P=0.009. To address the non-independence of the multiple pairwise Fst calculations, we applied a Bonferroni correction that resulted in an α-level of 0.0018, indicating that none of our Fst values is significantly different from 0. Our analysis of molecular variance also supported the lack of differentiation among breeding colonies and between live and dead birds. Differences among individuals within localities explained 100% of observed variance, whereas differences among breeding colonies and between live and dead birds failed to account for any fraction of the observed variance in our data. Mantel's test showed no correlation between genetic and geographical distances among the seven breeding colonies sampled (r2=0.08; P=0.127). These data, as well as the results of two multi-locus studies (Friesen and Piatt, 2003; Morris-Pocock et al., 2008), justify grouping all live birds into a single unit for the following analysis of mtDNA diversity and haplotype frequencies.
The MK test identified an excess of non-synonymous substitutions among polymorphic sites within species (Fisher's exact P=0.004) suggesting purifying selection. Tajima's D, Fu's Fs and R2 test values were significant for both species regardless of whether Common Murres were combined or divided into live and dead groups (Table 2), also suggesting either purifying selection or recent demographic expansion.
Although the Common Murre is a slightly more abundant species than the Thick-billed Murre in Alaska, 2.8 vs 2.2 × 106 (U.S. Fish and Wildlife Service, 2006a, 2006b), Hd and πn diversity were higher in Thick-billed than in Common Murres (Table 2). Dead Common Murres had greater Hd and πn than live Common Murres.
A single haplotype dominated each species. The frequency of the most common haplotype in the Thick-billed Murre was 43.5% followed by the second most common haplotype with frequency of 17.4%. In the Common Murre, the difference in frequency between the two most common haplotypes was much greater and exceeded an order of magnitude, 67.1% versus 6.4%, respectively (Figure 2). The proportion of the dominant haplotype was significantly lower among dead (59.1%; 95% CI 50.4–67.4%) Common Murres than among live ones (73.2%; 95% CI 66.3–79.9%; Fisher's exact P=0.01; Figure 1). The observed frequency of the most common haplotype among dead birds was below the 95% CI constructed under the null hypothesis of no difference between live and dead birds (65.4–81.1%). In fact, only 10 of our 10 000 simulated data sets had frequencies of the dominant haplotype ⩽59.1% (P<0.01). A traditional two-sample test of proportions, testing the equivalency between the proportion of the dominant haplotype for live and dead birds was also statistically significant (P=0.01). These results suggest that carriers of the dominant haplotype were significantly less likely to die during the mass die-off than carriers of other haplotypes.
The proportion of non-synonymous and synonymous substitutions among minor haplotypes did not differ significantly between dead (18:34) and live birds (10:35; Fisher's exact P=0.26). This non-significant result is unlikely to be simply due to the small sample sizes, and in fact a threefold increase of the sample sizes with the same proportions still results in a non-significant P-value (0.053). Therefore, during the die-off, non-synonymous substitutions in the mtDNA ND2 gene were not significantly more likely to be eliminated than synonymous substitutions.
Discussion
Recent multi-locus studies of the Common Murre phylogeography showed lack of geographical structuring and low mtDNA diversity across the North Pacific (Friesen and Piatt, 2003; Morris-Pocock et al., 2008). Our results are consistent with these findings. None of the pairwise Fst values were significantly different from 0, differences among breeding colonies failed to explain any proportion of the observed variation in data, and we found no correlation between geographical and genetic distances among breeding colonies. Our results also show that in Alaska the Common Murre has much lower mtDNA variation compared with that of the Thick-billed Murre (Figure 2). Both Hd and πn for the Thick-billed Murre were nearly twice that of the Common Murre (Table 2).
Our ND2 data also point to a generally non-neutral evolution of mtDNA in the Pacific Common Murre. All intraspecific neutrality tests (Tajima's D, Fu's Fs and R2) had significant values (Table 2). The MK test identified that the proportion of non-synonymous substitutions was significantly lower in fixed differences between species than in the intraspecific variation. Such differences usually are interpreted as evidence for purifying selection that is expected to preferentially eliminate non-synonymous substitutions through time (McDonald and Kreitman, 1991).
If the differences in haplotype frequencies between live and dead birds result from purifying selection the non-synonymous to synonymous substitutions ratio should differ between live and dead birds. However, we found no significant difference in the proportion of non-synonymous and synonymous substitutions among live and dead carriers of mutant haplotypes. This could indicate a random nature of the die-off. However, the comparison of haplotype frequencies between live and dead murres showed that although the same haplotype dominated both groups, its frequency was significantly higher among live than dead birds (Figure 1). These results are inconsistent with the effects of purifying selection or random elimination of haplotypes, rather they suggest a greater probability of survival through a die-off by carriers of the dominant haplotype, that is, with the positive selection on the dominant haplotype during die-offs.
The apparent disagreement between the results of the MK test and comparison of haplotype frequencies between live and dead murres is superficial and could be explained by the purifying and positive selection operating at different time scales. The MK test compares substitutions fixed during the course of evolution as the divergence of the two sister species to substitutions within both of these species. In other words, the MK test is based on the cumulative effect of selection affecting both sister species over a few million years as their divergence. The ND2 divergence of nearly 7% between the Common and Thick-billed murres corresponds to 1.5–3.5 million years depending on the molecular clock calibration used (Drovetski, 2003; Lovette, 2004; Arbogast et al., 2006). Therefore, the MK test provides no information on what kind of evolutionary forces could have affected one of the taxa during the few months of the die-off. During an occasional mass die-off, an estimated 10% of the Common Murre population is eliminated within a few months and this is likely a considerable underestimate (Piatt and van Pelt, 1997). Carriers of both non-synonymous and synonymous substitutions are impacted to a similar degree by a catastrophic, albeit short-term, event and have lower survival than the dominant haplotype. Three different statistical tests comparing haplotype frequencies between live and dead birds (Fisher's exact, P=0.01; bootstrapping, P<0.01 and normal approximation to the binomial distribution, P=0.01) identified this differential survival, which happens during die-offs. There is no reason to reject the possibility of occasional bursts of positive selection if the mtDNA is affected by long-term purifying selection.
Although our data show a significant difference in the probability of survival between carriers of the dominant and other haplotypes, it does not necessarily mean that selection operates directly on the mtDNA ND2 gene. Because mtDNA represents a single locus, the selection may be operating on any of the mtDNA genes. Furthermore, a number of cytonuclear interactions related to either production of nuclear-encoded proteins involved in the mitochondrial electron transport chain or to translation efficiency of mtDNA proteins can result in hitchhiking of the dominant ND2 haplotype.
Although differences in mtDNA haplotype frequencies between live and dead murres are necessary to identify the presence and type of selection, they are not sufficient. Fitness effects of small differences among mtDNA haplotypes have to be confirmed by independent data (Hughes, 2007). Experimental and field studies that assess differences in physiological performance between carriers of only slightly different mtDNA haplotypes under varying environmental conditions will be essential to validate our findings. This study provides guidance for such future investigations.
Data archiving
There were no data to deposit.
Accession codes
References
Ainley DG, Nettleship DN, Carter HR, Storey AE (2002). Common Murre (Uria aalge). In: Poole A (eds). The Birds of North America Online. Cornell Lab of Ornithology: Ithaca.
Akaike H (1974). A new look at the statistical model identification. IEEE Transact Automatic Control 19: 716–723.
Arbogast BS, Drovetski SV, Curry RL, Boag P, Grant PR, Grant RB et al. (2006). Origin and diversification of Galápagos mockingbirds. Evolution 60: 370–382.
Bailey EP, Davenport GH (1972). Die-off of Common Murres on the Alaska Peninsula and Unimak Island. Condor 74: 215–219.
Drovetski SV (2003). Plio-Pleistocene climatic oscillations, Holarctic biogeography and speciation in an avian subfamily. J Biogeogr 30: 1173–1181.
Drovetski SV, Zink RM IV F, Nesterov EV, EA K, Red'kin YA et al. (2004). Mitochondrial phylogeny of Locustella and related genera. J Avian Biol 35: 105–110.
Efron B, Tibshirani R (1986). Bootstrap methods for standard errors, confidence intervals, and other measures of statistical accuracy. Stat Sci 1: 54–77.
Efron B, Tibshirani R (1993). An Introduction to the Bootstrap. Chapman & Hall: Boca Raton, FL.
Excoffier L, Laval G, Schneider S (2005). Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evol Bioinform Online 1: 47–50.
Excoffier L, Smouse P, Quattro J (1992). Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131: 479–491.
Friesen VL, Piatt JF (2003). A genetic study to aid in restoration of murres, guillemots and murrelets to the Gulf of Alaska. Exxon Valdez Oil Spill Restoration Project Final Report (Restoration Project 00169). Alaska Biological Sciences Center, USGS: Anchorage, Alaska.
Fu Y-X (1997). Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147: 915–925.
Gromme OJ (1927). Some highlights on the faunal life of the Alaskan Peninsula. Milwaukee Public Museum Yearbook 7: 30–45.
Hackett SJ (1996). Molecular phylogenetics and biogeography of tanagers in the genus Ramphocelus (Aves). Mol Phylogenet Evol 5: 368–382.
Hughes A (2007). Looking for Darwin in all the wrong places: the misguided quest for positive selection at the nucleotide sequence level. Heredity 99: 364–373.
Librado P, Rozas J (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25: 1451–1452.
Lovette IJ (2004). Mitochondrial dating and mixed support for the ‘2% rule’ in birds. Auk 121: 1–6.
Mantel N (1967). The detection of disease clustering and a generalized regression approach. Cancer Res 27: 209–220.
McDonald JH, Kreitman M (1991). Adaptive protein evolution at the Adh locus in Drosophila. Nature 351: 652–654.
Morris-Pocock JA, Tylor SA, Birt TP, Damus M, Piatt JF, Warheit KI et al. (2008). Population genetic structure in Atlantic and Pacific Ocean Common Murres (Uria aalge): natural replicate tests of post-Pleistocene evolution. Mol Ecol 17: 4859–4873.
Piatt JF, van Pelt TI (1997). Mass-mortality of Guillemots (Uria aalge) in the Gulf of Alaska in 1993. Mar Pollut Bull 34: 656–662.
Posada D (2008). jModelTest: phylogenetic model averaging. Mol Biol Evol 25: 1253–1256.
Ramos-Onsins SE, Rozas J (2002). Statistical properties of new neutrality tests against population growth. Mol Biol Evol 19: 2092–2100.
Swofford DL (1998). PAUP*: Phylogenetic Analysis Using Parsimony (* and Other Methods), 4.0 ed. Sinauer: Sunderland, MA.
Tajima F (1989). Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595.
Tajima F (1996). The amount of DNA polymorphism maintained in a finite population when the neutral mutation rate varies among sites. Genetics 143: 1457–1465.
US Fish and Wildlife Service (2006a). Common Murre (Uria aalge) Alaska Seabird Information Series. US Fish and Wildlife Service: Alaska.
US Fish and Wildlife Service (2006b). Thick-billed Murre (Uria lomvia) Alaska Seabird Information Series. US Fish and Wildlife Service: Alaska.
Vader W, Barrett RT, Erikstad KE, Strann K-B (1990). Differential responses of Common and Thick-Billed Murres to a crash in the capelin stock in the southern Barents Sea. Stud Avian Biol 14: 175–180.
Zar JH (1996). Biostatistical Analysis, 3rd edn. Prentice Hall: Upper Saddle River, New Jersey, USA. 662pp.
Acknowledgements
We are grateful to Stuart Baird, Scott A Hatch, Robert M Suryan, Sandra L Talbot and two anonymous reviewers for helpful comments and suggestions. This study was supported by grants from the EVOS Trustees Council and the North Pacific Research Board (award #320 to ASK).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Drovetski, S., Kitaysky, A., Mode, N. et al. mtDNA haplotypes differ in their probability of being eliminated by a mass die-off in an abundant seabird. Heredity 109, 29–33 (2012). https://doi.org/10.1038/hdy.2012.4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2012.4
