
Abstract
Developmental eye diseases, including cataract/microcornea, Peters anomaly and coloboma/microphthalmia/anophthalmia, are caused by mutations encoding many different signalling and structural proteins in the developing eye. All modes of Mendelian inheritance occur and many are sporadic cases, so provision of accurate recurrence risk information for families and affected individuals is highly challenging. Extreme genetic heterogeneity renders testing for all known disease genes clinically unavailable with traditional methods. We used whole-exome sequencing in 11 unrelated developmental eye disease patients, as it provides a strategy for assessment of multiple disease genes simultaneously. We identified five causative variants in four patients in four different disease genes, GJA8, CRYGC, PAX6 and CYP1B1. This detection rate (36%) is high for a group of patients where clinical testing is frequently not undertaken due to lack of availability and cost. The results affected clinical management in all cases. These variants were detected in the cataract/microcornea and Peters anomaly patients. In two patients with coloboma/microphthalmia, variants in ABCB6 and GDF3 were identified with incomplete penetrance, highlighting the complex inheritance pattern associated with this phenotype. In the coloboma/microphthalmia patients, four other variants were identified in CYP1B1, and CYP1B1 emerged as a candidate gene to be considered as a modifier in coloboma/microphthalmia.
Similar content being viewed by others


INTRODUCTION
Developmental eye diseases are those where development of the eye can be affected through a number of different processes influencing development of the various components of the eye, including the cornea, lens, iris and optic fissure. Progress in genetic diagnostic capability in patients with developmental eye diseases, including cataract/microcornea, disorders of the anterior segment and coloboma/microphthalmia/anophthalmia, has been hampered by the marked genetic heterogeneity of these conditions. Although a number of disease genes have been identified, pursuit of a molecular diagnosis is often not clear-cut as there is an overlap in the clinical features associated with mutations in the various underlying disease genes (Figure 1).1, 2 In some cases, molecular diagnosis may be aided by syndrome identification or a chromosomal anomaly may be revealed.3 For isolated cases, which are the majority, molecular diagnosis is challenging, as the required sequential sequencing for identification of underlying mutation/s is rarely completed owing to the lack of an available panel covering all the known genes.

Genetic heterogeneity and overlap in phenotypes of developmental eye disorders. Developmental eye phenotypes are shown, and as indicated, variants in many of the genes can cause different types of developmental eye phenotypes. Normal font of the gene indicates causation of an eye disease phenotype without systemic abnormality, while bold font of the gene indicates eye disease with additional systemic/syndromic features.
Congenital cataracts associated with microcornea, with or without microphthalmia, may be caused by mutations in at least nine genes, including GJA8, CRYAA, CRYBB1, CRYBA4, CRYBB2, CRYGC, CRYGD, MAF and FOXE3.4 Other disorders of the anterior segment of the eye also show marked genetic heterogeneity and include isolated trabecular meshwork abnormality or primary congenital glaucoma, as well as iris abnormalities, including Axenfeld and Rieger anomalies, and corneal dysgenesis or Peters anomaly. In Peters anomaly, there are reports identifying mutations in a number of genes, including PAX6, PITX2, FOXC1, MAF, FOXE3, PITX3 and CYP1B1.1, 5, 6 Overlap in clinical features is evident with some patients with mutations in a particular gene having cataract/microcornea while others may have a specific anterior segment dysgenesis phenotype, and microphthalmia may also be present.7 As an example, mutations in the gene FOXE3 may be found in patients with cataract, disorders of the anterior segment and microphthalmia (Figure 1).8, 9, 10
The developmental ocular anomaly of coloboma, due to delay in closure of the optic fissure, may affect the iris, choroid, retina and/or optic disc.11 It is frequently associated with microphthalmia or anophthalmia.12 In patients with microphthalmia and anophthalmia, attempts have been made to stratify patients on the basis of the presence of associated eye malformations; however, the overlap in phenotypic features makes this difficult (Figure 1).13, 14
In this study, we applied exome capture and sequencing in patients with developmental eye diseases, including cataract/microcornea, Peters anomaly and coloboma/microphthalmia/anophthalmia, and analysed the 56 genes currently known to be implicated in developmental eye disease (Supplementary Table S1).
MATERIALS AND METHODS
Patients
Eleven probands with developmental eye disease, four with cataract/microcornea, three with Peters anomaly and four with coloboma/microphthalmia, were chosen for the study (Table 1). Probands were selected where there were parental and/or other family member samples available so that segregation analysis could subsequently be performed (Figure 2). Probands and family members underwent full ophthalmic examination (Supplementary Table S2). All the four probands (Patients 1–4, Table 1) in the cataract and microcornea/microphthalmia group had severe bilateral eye disease that had been identified soon after birth. One proband (Patient 4, I.2 in Family 4) also had multiple iris processes adherent to the lens, highlighting that while the main ocular phenotype is recorded as cataract in this case, there can be associated anterior segment anomalies in these patients complicating decisions regarding candidate disease genes to be studied. In the three probands (Patients 5–7, Table 1) with a predominant Peters anomaly phenotype, all patients had bilateral corneal opacities and were diagnosed soon after birth. Two also had lens abnormalities, one with anterior polar lens opacities (Patient 5, II.1 in Family 5) and the other with rudimentary lens (Patient 6, II.1 in Family 6), and two also had microphthalmia (Patient 6, II.1 in Family 6, and Patient 7, II.2 in Family 7). All the four probands in the coloboma/microphthalmia group (Patients 8 to 11, Table 1) had bilateral eye disease detected at or soon after birth, and one of these also had anterior polar cataracts (Patient 8, II.1 in Family 8). In view of the overlap in phenotypic features that may occur in these groups of developmental eye conditions and the large number of candidate disease genes, we decided to take a whole-exome sequencing approach followed by analysis of known disease genes in these disorders. Genomic DNA was isolated from leukocytes of peripheral venous blood. All experiments were performed in accordance with the ethical tenets of The Children’s Hospital at Westmead, Sydney, NSW, Australia.

Pedigrees of the families studied. A proband from each of the families underwent exome sequencing. Probands are indicated by arrows. Asterisk indicates DNA sample available. Filled squares or circles indicate affected individuals.
Exome sequencing, assembly and variant calling
Whole-exome sequence capture was performed in a proband from each of the 11 families. For Patient 1, this was performed using the TruSeq exome enrichment system (Illumina Inc., San Diego, CA, USA), and for all other probands the SeqCap EZ Human Exome Library v3.0 (Roche NimbleGen, Inc., Madison, WI, USA) was used. Sequencing was performed on the Illumina HiSeq 2000 using 100 bp paired-end read sequencing protocol (Axeq Technologies Asia, Macrogen Inc., Seoul, South Korea and at the Genome Institute of Singapore for Patient 4). Reads were aligned to the reference human genome (hg19) using the Burrows–Wheeler alignment tool15 using default parameters. Reads with a mapping quality score ≤10 were discarded using SAMtools16 and the Picard MarkDuplicates tool was used to identify and discard read duplicates (http://picard.sourceforge.net).
SNPs and indels were identified using the Genome Analysis Toolkit (GATK) version 1.6-9,17 except for Patient 4 where CASAVA 1.8 (Illumina Inc.) was used. Variants in 56 known developmental eye disease genes (Supplementary Table S1) were annotated using Annovar.18 Known polymorphisms were identified using dbSNP132, dbSNP135, 1000 genomes (30 April 2012 release) and NHLBI exome project (ESP5400 release), and minor allele frequencies were recorded from 1000 genomes and ESP5400. Minor allele frequencies were recorded manually from dbSNP when required. Coding sequence variants were annotated with prediction of pathogenicity based on the SIFT,19 PolyPhen-220 and MutationTaster21 algorithms using the pre-calculated data provided in dbNSFP22 (Supplementary Figure S1).
Variant prioritisation
Variants within the 56 known eye development disease genes (Figure 1, Supplementary Table S1) were selected for further analysis when all of the following conditions were met as in previous similar approaches23 (Supplementary Figure S1). Variants that were nonsense, frameshift or canonical splice site variants, affecting the two nucleotides immediately adjacent to the splice donor or acceptor sites, were considered to be pathogenic. We used a classification system for missense variants using: (1) missense prediction software assessing pathogenicity at the amino-acid level and (2) evolutionary conservation at the nucleotide level.24 We used the missense prediction programs, PolyPhen-2, SIFT and MutTaster, and the different scores from these tools were derived according to the rules described by Liu et al.22 Results from the three different tools were combined to give a majority vote resulting in a single classification as ‘damaging’ or ‘tolerated’. For the classification based on evolutionary conservation, all variants with a phylo P>0.95 were considered conserved (C), otherwise non-conserved (NC). Hence, missense variants were considered ‘probably pathogenic’ when the population frequency was <1% in the public databases as described above, and if either the prediction programs or conservation tool predicted a damaging or highly conserved allele. Otherwise the variant was considered ‘probably benign’. All ‘probably pathogenic’ alleles were subjected to segregation analysis. A cutoff of <1% population frequency in the control databases was used as isolated developmental eye disorders are rare,25 and in addition, each disease gene accounts only for a small proportion of cases.
Variant validation and segregation analysis
Using this approach, between 0 and 4 variants per sample were selected for validation leading to a total of 16 variants in the 11 probands (Table 1, Supplementary Table S3). Variants were validated using the conventional Sanger sequencing. Reference sequences used were: GJA8, NM_005267.4; CRYGC, NM_020989.3; CYP1B1, NM_000104.3; PAX6, NM_000280.4; CRYBA1, NM_005208.4; GCNT2, NM_145655.3; BFSP1, NM_001161705.1; ABCB6, NM_005689.2; SLC16A12, NM_213606.3; and GDF3, NM_020634.1. Primers for Sanger sequencing are available upon request. DNAs from additional family members were sequenced to enable segregation analysis, and proteins and variants were examined for previous disease reports. Variants identified in GJA8, CRYBA1, CRYGC, BFSP1, GCNT2 and PAX6 genes were submitted to Eye diseases—Leiden Open Variation Databases (LOVD) http://grenada.lumc.nl/LOVD2/eye/home.php and http://lsdb.hgu.mrc.ac.uk/home.php?select_db=PAX6. Other variants were submitted to the ClinVar database (www.ncbi.nlm.nih.gov/clinvar/).
RESULTS
Exome and target region analysis
In the 11 developmental eye disease probands, whole-exome sequencing resulted in an average of 59 311 192 reads/sample. The mean depth of coverage of the targeted regions across the exome was 43X across all samples, with on average 94% of all targeted regions being covered by ≥10X reads (Supplementary Table S3). For the 56 developmental eye disease genes across the 11 samples, 87.8–95.6% of the coding regions were covered by ≥5X reads.
After selection for exonic and canonical splice site variants across the 56 genes in the 11 samples, 36–55 variants were identified per sample (Supplementary Table S3). After application of our systematic variant prioritisation tool (Supplementary Figure S1), 16 predicted pathogenic variants were identified. Sanger sequencing confirmed these variants. The variants confirmed in probands were further sequenced in parental and sibling samples where available.
Five causative variants identified in cataract/microcornea and Peters anomaly patients
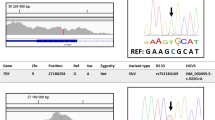
In two of four cataract/microcornea patients, (Table 1) two pathogenic variants with appropriate segregation were identified, and one of these was novel. In Patient 1 (Family 1, II.1), we detected a heterozygous mutation in gap junction alpha-8 (GJA8), c. 593G>A, predicting a deleterious substitution of arginine by glutamine (p.(Arg198Gln)), which segregated with disease in the family (Table 2, Figure 3a, Supplementary Figure S2), and is previously reported in autosomal-dominant cataract/microcornea.26 In Patient 4 (Family 4, II.1), a novel heterozygous variant was detected in CRYGC, c.497C>T, p.(Ser166Phe). This predicted deleterious variant affected a highly conserved amino acid in the second beta/gamma crystallin domain of this protein, was not present in the 1000 genome, ESP5400 or dbSNP databases and segregated with disease in the affected family members (Table 2, Figures 3b and e, Supplementary Figure S2).

Pedigrees and clinical images in families with pathogenic variants and segregation indicating autosomal dominant or recessive inheritance and alignment of amino-acid sequences for two novel mutations. (a) Family 1 pedigree and nuclear cataract in patient III.1. Mutation in GJA8 c. 593G>A, p.(Arg198Gln), de novo autosomal-dominant mutation in II.1 (Patient 1) transmitted to III.1, III.2 and III.3, (b) Family 4 pedigree and nuclear cataract in patient II.1. Mutation in CRYGC, c.497C>T, p.(Ser166Phe), autosomal-dominant mutation in I.2 (Patient 4), transmitted to II.1 and II.2, (c) Family 5 pedigree and anterior polar lens opacity in patient II.1(Patient 5) with Peters anomaly post-penetrating keratoplasty. Compound heterozygous mutations in II.1 in CYP1B1, c.171G>A, (p.Trp57*), inherited from unaffected father, I.1, and c.1200_1209dup, p.(Thr404Serfs*30) inherited from the unaffected mother, I.2, (d) Family 6 pedigree. De novo autosomal-dominant mutation in patient, II.1, (Patient 6) in PAX6, c.152G>T, p.(Gly51Val). (e) Alignment of amino-acid sequences for the two novel mutations, CRYGC, p.(Ser166Phe) and PAX6, p.(Gly51Val). The reference sequences for CRYGC and PAX6 correspondingly: Homo sapiens NP_066269.1 and NP_000271.1, Mus musculus NP_031801.1 and NP_001231129.1, Rattus norvegicus NP_264077.1 and NP_037133.1, Bos taurus NP_001013613.1 and NP_001035735.1, Danio rerio NP_001018630.1 and NP_571716.1, and X.tropicalis XP_002937158.1 and NP_001006763.1.
Examination of exome sequence data from three individuals with Peters anomaly led to the identification of causative variants in two patients(Table 1) In Patient 5 (Family 5, II.1), two heterozygous variants in CYP1B1 (p.(Thr404fs), p.(Trp57*)) were identified. One of the variants was a 10 nucleotide insertion, c.1200_1209dupTCATGCCACC, which resulted in a frameshift mutation (p.(Thr404Serfs*30)) leading to a shortened abnormal protein. This same variant has been reported in primary congenital glaucoma,27 as part of a compound mutation, but not previously in Peters anomaly. The second heterozygous CYP1B1variant in this patient, c.171G>A, predicted a premature stop codon (p.(Trp57*)) (Table 2, Figure 3c, Supplementary Figure S2) and was reported previously in Peters anomaly.6
In Patient 6 (Family 6, II.1) with Peters anomaly, we detected a novel heterozygous variant in PAX6 leading to a missense change in a highly conserved glycine in the paired box domain (c.152G>T, p.(Gly51Val)). This was predicted as a pathogenic variant due to a high conservation score and damaging scores using missense prediction tools, and this variant has not been identified in any control panels. Sanger sequencing showed that this was a de novo heterozygous variant in the patient, with absence of the mutation in the unaffected parents (Table 2, Figures 3d and e, Supplementary Figure S2).
Two other variants were prioritised in the cataract/microcornea patients. One of these, in Patient 4 (Family 4, I.2), was not considered further as it was a variant in an isoform of GCNT2, c.505G>A, p.(Ala169Thr), (rs56106312, Reference sequence: NM_145655.3), that is not expressed in lens and hence not associated with cataract.28 In Patient 3 (Family 3, II.1) with cataract/microcornea, a heterozygous variant in CRYBA1, c.475G>A, predicted a probable pathogenic variant, p.(Gly159Ser), (rs117757092, frequency in controls ESP5400, 0.00316 and 1000 genomes, 0.0009), but this was also found in the patient’s unaffected mother (Table 3). Variable penetrance and expression occur in developmental eye diseases and are reported in cataract/microcornea patients, including those with mutations in crystallin genes.29 Whether this variant may contribute to this patient’s phenotype will be determined as more variants in these genes are identified through broader sequencing strategies in patient cohorts made available by Next-Generation Sequencing (NGS).
Variants with incomplete penetrance in coloboma/microphthalmia patients
A number of variants were prioritised in three out of four of our coloboma/microphthalmia patients (Table 1), and we first considered genes previously reported as known disease genes particularly in this disorder (Figure 1). Patient 11 (Family 11, II.1), with microphthalmia and coloboma, was heterozygous for a novel missense variant in GDF3, c. 974C>T, p.(Pro325Leu). This variant affects a conserved residue in the TGF-beta domain of this protein and is predicted to be a damaging variant using Polyphen-2, SIFT and Mutation Taster (Table 3). This patient was of Indian ethnicity, and the variant was not present in ESP5400 or 1000 genome controls, including 70 individuals of South Asian origin. This variant was also present in the patient’s unaffected father (Table 3, Supplementary Figure S3). Other patients with microphthalmia and coloboma have been found to have heterozygous missense variants in the TGF-beta domain of GDF3, and variable penetrance has been previously noted.30
In Patient 10 (Family 10, II.1), we detected a rare heterozygous variant in ABCB6, c.575G>A, p.(Arg192Gln) (Table 3, Supplementary Figure S3). ABCB6 encodes a protein responsible for porphyrin transport and is located in the outer mitochondrial membrane. A missense variant in this gene has been found to segregate in an autosomal-dominant manner in a family with coloboma, and another heterozygous missense variant was found in three isolated cases. Results from zebrafish rescue experiments indicated that these variants lead to loss of protein function.31 However, in two more recent studies, several individuals homozygous for deleterious loss of function mutations, including frameshift and nonsense mutations, were found to have a newly identified rare Lan blood group antigen. There were no reports of coloboma or eye defects in individuals homozygous or heterozygous for these variants 32. In our family (Family 10), the unaffected father also carried ABCB6, c.575G>A (p.(Arg192Gln)) in the heterozygous state, indicating that this is a variant with incomplete penetrance and that other factor/s are contributing to the disease phenotype in Patient 10.
Variants in developmental eye disease genes across the cohort
As there can be overlap in clinical features in patients with developmental eye disorders, we considered prioritised variants from known developmental eye disease genes across the 11 probands with developmental eye disease. This revealed one novel and three rare variants in CYP1B1 in two out of four coloboma/microphthalmia patients (Table 1). In Patient 9 (Family 9, II.1), there was a novel heterozygous duplication in CYP1B1, c.868dup, predicting a shortened protein with the normal amino-acid sequence finishing at amino acid 290, p.(Arg290Profs*37), compared with the normal 543 amino-acid protein. This variant was not present in any of the control databases, was present in the patient’s unaffected mother but was not present in her more mildly affected brother (Table 3, Supplementary Figure S3). Patient 9 also had a rare missense mutation in CYP1B1, c.241T>A, p.(Tyr81Asn), which was present in her affected brother, her unaffected father but not her unaffected mother (Table 3, Supplementary Figure S3). This missense mutation is in a conserved residue in the P450 domain of this protein (http://smart.embl.de), and previous enzymatic studies have indicated that it is a hypomorphic allele.33 Patient 11 (Family 11, II.1) who had a heterozygous change in GDF3, was also a compound heterozygote for two missense mutations in CYP1B1, c.1103G>A, p.(Arg368His), and c.685G>A, p.(Glu229Lys) (Table 3, Supplementary Figure S3). Both of these variants have been found to have higher frequencies in individuals with primary congenital glaucoma than in control populations, including in controls with this patient’s ethnicity. Enzymatic assays indicate c.1103G>A, p.(Arg368His) has markedly reduced activity while c.685G>A, p.(Glu229Lys) is a hypomorphic allele.33, 34 There was no evidence of glaucoma or Peters anomaly in Patient 9 (Family 9, II.1) or Patient 11 (Family 11, II.1). CYP1B1 can contribute to retinoic acid synthesis in embryonic development,35 and retinoic acid receptor signalling regulates choroid fissure closure.36 The presence of variation in two genes, GDF3 and CYP1B1, which may modulate choroid fissure closure, may be relevant as contributing factors to the disease in Patient 11 (Family 11, II.1).
Patients 9 (Family 9, II.1) and 11 (Family 11, II.1), both with coloboma/microphthalmia, also had rare heterozygous variants in BFSP1, which encodes a beaded filament lens protein. Patient 9, in addition to her variants in CYP1B1 described above, had a 2-bp deletion in BFSP1 that changed the stop codon to a lysine, resulting in the addition of seven random amino acids to the end of the protein, c.1620_1621del, p.(*541Lysext*7). For Patient 9, this variant was also present in her affected sib, as well as her unaffected mother (Table 3, Supplementary Figure S3). Patient 11, in addition to his variants in GDF3 and CYP1B1 described above, also had a variant in BFSP1, which was a missense change in a conserved amino acid in the coiled coil domain of the protein, c.401G>C, p.(Cys134Ser). In Patient 11’s family, the BFSP1 variant was present in heterozygous form in both his parents (Table 3, Supplementary Figure S3).
Patient 10 (Family 10, II.1), also with coloboma/microphthalmia, in addition to his variant in ABCB6 described above, also had a rare heterozygous variant in SLC16A12, c.472T>C, p.(Ser158Pro) (Table 3, Supplementary Figure S3), a cataract disease gene previously identified in an autosomal-dominant cataract family.37 This variant was in a conserved predicted transmembrane domain of the protein and was also present in Patient 10’s unaffected mother. Patients with mutations in other lens-related genes are reported with coloboma, as lens factors may influence anterior segment development.1 Hence, while these BFSP1 and SLC16A12 variants are not causative variants by themselves, as they are present in unaffected parents, it is possible that they may be modifying factors in coloboma/microphthalmia, and observation for variations in these and similar genes is warranted in future exome and genome-wide studies of other coloboma/microphthalmia patients.
DISCUSSION
In this study, we developed an investigation tool for developmental eye disease, consisting of exome sequencing of patients, followed by systematic analysis of the known disease genes in the disorders under study and interpretation of all detected genetic variants across the cohort. The clinical utility of this approach is highlighted by the diagnostic yield in our sporadic cases, revealing both autosomal recessive and de novo dominant mutations. The presence of de novo dominant mutations confers a significant risk for transmitting the disease to the patient’s offspring. The presence of compound heterozygous mutations confers a significant recurrence risk for the patient’s parents. These findings illustrate the molecular diagnostic power of exome capture followed by targeted analysis in these heterogeneous eye disorders and also the huge impact of this method for affected families.
The strength of our approach lies in the parallel analysis of all known eye developmental disease genes in our patients with unknown molecular cause of disease. Systematic variant prioritisation reduced the initially high number of identified variants (∼45 per patient) by 97% to a number that is manageable to validate by Sanger sequencing (approximately 1.4 variants per patient). Using a systematic approach for assessment of pathogenicity of the validated variants, our NGS approach resulted in a clear molecular diagnosis in 4 out of 11 patients, which is an encouraging detection rate in a group of diseases where molecular diagnosis is often not undertaken because of low detection rates in each of the genes which, up until now, have required individual sequential sequencing.
Several individuals in our cohort were the first affected person in their family, where there were concerns about recurrence risk expressed by family members and the individuals themselves. Cataract associated with microcornea is usually an autosomal-dominant condition, although autosomal-recessive cases are reported.38 With sequential Sanger sequencing of several genes in research cohorts, the mutation detection rate is in the vicinity of 10–50%.4, 39 Such testing is not possible to be undertaken in the clinical setting, because no panels of these genes are available for diagnostic screening. Using our approach, we have detected a novel heterozygous pathogenic mutation in CRYGC in Patient 4 and a known pathogenic variant in GJA8 in Patient 1. In both the families, infantile cataract management has been complicated by secondary glaucoma and requirement for repeated surgical procedures and impaired vision in all cases. Provision of molecular genetic diagnosis significantly improves the genetic information and options available for these families.
Peters anomaly is a severe disorder affecting vision that is extremely genetically heterogeneous with autosomal-dominant and -recessive forms described. In two out of the three isolated cases in our cohort, we have identified disease-causing mutations in two different genes. In Patient 5, two previously reported mutations in CYP1B1 were present. One of the mutations had been reported in Peters anomaly while the other was in a patient with primary congenital glaucoma. It has been previously noted that mutations in CYP1B1 can lead to Peters anomaly or primary congenital glaucoma, and this suggests that they may have the same etiology due to the abnormal migration of neural crest cells.40 Each of the unaffected parents was found to be heterozygous for one of the variants. Before this information, the parents were given a recurrence risk varying from <1% (new autosomal-dominant condition) to 25% (autosomal-recessive condition). Molecular diagnosis in the proband clarifies the recurrence risk to 25% for the parents, and the transmission risk for the affected individual is low.
In Patient 6 with Peters anomaly, a novel variant in PAX6 has been identified. This is in the paired domain and is predicted to be a damaging variant with high conservation across species. Neither of the unaffected parents has this change. For this couple, the recurrence risk has now been clarified to <1% on the basis that this is a new autosomal-dominant change in the patient. However, for the affected individual, the transmission risk is 50%. Peters anomaly is frequently seen as an isolated condition, and provision of recurrence and transmission risk information lacks clarity in the absence of a molecular diagnosis.
Molecular detection rates in any of the possible known genes in patients with coloboma/microphthalmia have been generally very low in those patients who do not have associated anophthalmia in the contralateral eye. This has been confirmed in this study where a combination of pathogenicity assessment and segregation analysis has not found a primarily pathogenic gene in any of the coloboma/microphthalmia patients. Patient 11 has a heterozygous probably pathogenic mutation in GDF3, which is also present in his unaffected father, and while variations in this gene have been noted in association with microphthalmia and coloboma, variability in penetrance has also been described. This patient is also a compound heterozygote for two probably pathogenic missense variants in CYP1B1. Deleterious homozygous or compound heterozygous mutations in CYP1B1 are found in patients with primary congenital glaucoma and Peters anomaly, and CYP1B1 may also regulate retinoic acid signalling for proper optic fissure closure. It is possible that variation in both GDF3 and CYP1B1 may be contributing to Patient 11’s phenotype of coloboma and microphthalmia. Interestingly, one other microphthalmia/coloboma patient, Patient 9, had a heterozygous frameshift variant in CYP1B1. This was also present in her unaffected mother and was not present in her affected brother who had a less severe phenotype with only a subtle retinal coloboma revealed by fundal examination. This suggests that Patient 9 and her more mildly affected brother may have pathogenic variation in an as yet undiscovered coloboma/microphthalmia disease gene and that the disease severity may be modulated by the presence of the frameshift variant in CYP1B1 in Patient 9. This marks CYP1B1 as a candidate to be considered as a modifier gene in other coloboma/microphthalmia cases.
In the whole-exome sequencing approach we undertook in the 11 probands in our study, we achieved a mean depth of coverage of the targeted regions across the exome of 43X across all samples. In addition, there was incomplete coverage, as for the 56 developmental eye disease genes across the 11 samples, 87.8–95.6% of the coding regions were covered by ≥5X reads. The lack of coverage was identified particularly in GC-rich and repetitive regions. Ideally, to maximise the likelihood of mutation variant detection, a higher depth of coverage and capture in all of the possible disease-causing regions would be preferred. Nevertheless, when we compared detection rates from this study, with previous research studies involving conventional sequencing techniques to screen mutations in genes known to be associated with developmental eye diseases, we found comparable detection rates. Sanger sequencing studies have shown a detection rate of approximately 10–50% in patients with cataract/microcornea,4, 39 and our approach has resulted in the identification of a clear-cut disease causing variant in two out of four of our cataract/microcornea patients. For the Peters anomaly group, there are mostly isolated case reports of mutations in different genes for these patients, although in one study there were four genes investigated, and clear-cut mutations were identified in 20% (3/15) of patients.41 In patients with microphthalmia/coloboma, detection rates for clear-cut pathogenic variants are generally very low, in the vicinity of 1–2% or less as found in our study.13, 42, 43
A targeted NGS strategy of the known disease genes could possibly lead to a higher detection rate, as a higher depth of coverage can be achieved with this. However, where such a strategy was undertaken in the severe anophthalmia/microphthalmia group, where there is a known higher detection rate, no disease gene was identified in almost one-third of the patients.44 This suggests that there are still a significant proportion of unknown disease genes in patients with developmental eye disease. The approach we followed provides an option for a mutation detection route for at least a proportion of patients with these conditions, as service laboratories are now offering whole-exome sequencing followed by an analysis of a target list of genes. Any likely pathogenic variants can be confirmed by Sanger sequencing, and segregation can be confirmed in family members. As strategies for capture of GC-rich and repetitive regions improve, and increased depth of coverage can be undertaken for less cost in the future, it is likely that detection rates will improve. For patients where no disease gene variant has been identified and with suitable family structure and availability of samples for analysis, research studies may be able to identify novel disease genes.
This paper presents an exome-sequencing strategy for disease gene identification in genetically heterogeneous developmental eye diseases, including cataract/microcornea, Peters anomaly and coloboma/microphthalmia. Overall, we identified causative mutations in known disease genes, GJA8, CRYGC, CYP1B1 and PAX6, in 36% (4/11) of families, an encouraging detection rate for diseases where molecular genetic diagnosis is frequently not undertaken owing to the high heterogeneity and lack of available panels for clinical diagnosis. These results provided clinically useful information in all cases. Clear-cut detection rates were better for the cataract/microcornea (2/4 families) and Peters anomaly (2/3 families) groups, compared with the coloboma/microphthalmia (0/4 families) group. Variants associated with variable penetrance were identified in CRYBA1 in the cataract/microcornea group and ABCB6 and GDF3 in the coloboma/microphthalmia group. Our broad strategy in disease gene examination also identified CYP1B1 as a new potential modifier in coloboma/microphthalmia. Our results support the high heterogeneity among patients with developmental eye diseases and show that the exome-sequencing strategy can be an expeditious approach for the identification of disease-causing and modifier variants in these conditions.
References
Jamieson RV, Perveen R, Kerr B et al: Domain disruption and mutation of the bZIP transcription factor, MAF, associated with cataract, ocular anterior segment dysgenesis and coloboma. Hum Mol Genet 2002; 11: 33–42.
Semina EV, Ferrell RE, Mintz-Hittner HA et al: A novel homeobox gene PITX3 is mutated in families with autosomal-dominant cataracts and ASMD. Nat Genet 1998; 19: 167–170.
Nolen LD, Amor D, Haywood A et al: Deletion at 14q22-23 indicates a contiguous gene syndrome comprising anophthalmia, pituitary hypoplasia, and ear anomalies. Am J Med Genet A 2006; 140: 1711–1718.
Hansen L, Yao W, Eiberg H et al: Genetic heterogeneity in microcornea-cataract: five novel mutations in CRYAA, CRYGD, and GJA8. Invest Ophthalmol Vis Sci 2007; 48: 3937–3944.
Hanson IM, Fletcher JM, Jordan T et al: Mutations at the PAX6 locus are found in heterogeneous anterior segment malformations including Peters’ anomaly. Nat Genet 1994; 6: 168–173.
Vincent A, Billingsley G, Priston M et al: Phenotypic heterogeneity of CYP1B1: mutations in a patient with Peters' anomaly. J Med Genet 2001; 38: 324–326.
Summers KM, Withers SJ, Gole GA, Piras S, Taylor PJ : Anterior segment mesenchymal dysgenesis in a large Australian family is associated with the recurrent 17 bp duplication in PITX3. Mol Vis 2008; 14: 2010–2015.
Reis LM, Tyler RC, Schneider A et al: FOXE3 plays a significant role in autosomal recessive microphthalmia. Am J Med Genet A 2010; 152A: 582–590.
Doucette L, Green J, Fernandez B, Johnson GJ, Parfrey P, Young TL : A novel, non-stop mutation in FOXE3 causes an autosomal dominant form of variable anterior segment dysgenesis including Peters anomaly. Eur J Hum Genet 2011; 19: 293–299.
Bremond-Gignac D, Bitoun P, Reis LM, Copin H, Murray JC, Semina EV : Identification of dominant FOXE3 and PAX6 mutations in patients with congenital cataract and aniridia. Mol Vis 2010; 16: 1705–1711.
Gregory-Evans CY, Williams MJ, Halford S, Gregory-Evans K : Ocular coloboma: a reassessment in the age of molecular neuroscience. J Med Genet 2004; 41: 881–891.
Verma AS, FitzPatrick DR : Anophthalmia and microphthalmia. Orphanet J Rare Dis 2007; 2: 47.
Bakrania P, Efthymiou M, Klein JC et al: Mutations in BMP4 cause eye, brain, and digit developmental anomalies: overlap between the BMP4 and hedgehog signaling pathways. Am J Hum Genet 2008; 82: 304–319.
Mihelec M, Abraham P, Gibson K et al: Novel SOX2 partner-factor domain mutation in a four-generation family. Eur J Hum Genet 2009; 17: 1417–1422.
Li H, Durbin R : Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009; 25: 1754–1760.
Li H, Handsaker B, Wysoker A et al: The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009; 25: 2078–2079.
McKenna A, Hanna M, Banks E et al: The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010; 20: 1297–1303.
Wang K, Li M, Hakonarson H : ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010; 38: e164.
Ng PC, Henikoff S : Predicting deleterious amino acid substitutions. Genome Res 2001; 11: 863–874.
Adzhubei IA, Schmidt S, Peshkin L et al: A method and server for predicting damaging missense mutations. Nat Methods 2010; 7: 248–249.
Schwarz JM, Rodelsperger C, Schuelke M, Seelow D : MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 2010; 7: 575–576.
Liu X, Jian X, Boerwinkle E : dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions. Hum Mutat 2011; 32: 894–899.
de Ligt J, Willemsen MH, van Bon BW et al: Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med 2012; 367: 1921–1929.
Neveling K, Collin RW, Gilissen C et al: Next-generation genetic testing for retinitis pigmentosa. Hum Mutat 2012; 33: 963–972.
Bermejo E, Martinez-Frias ML : Congenital eye malformations: clinical-epidemiological analysis of 1,124,654 consecutive births in Spain. Am J Med Genet 1998; 75: 497–504.
Devi RR, Vijayalakshmi P : Novel mutations in GJA8 associated with autosomal dominant congenital cataract and microcornea. Mol Vis 2006; 12: 190–195.
Stoilov I, Akarsu AN, Alozie I et al: Sequence analysis and homology modeling suggest that primary congenital glaucoma on 2p21 results from mutations disrupting either the hinge region or the conserved core structures of cytochrome P4501B1. Am J Hum Genet 1998; 62: 573–584.
Yu LC, Twu YC, Chou ML et al: The molecular genetics of the human I locus and molecular background explain the partial association of the adult i phenotype with congenital cataracts. Blood 2003; 101: 2081–2088.
Andley UP : Effects of alpha-crystallin on lens cell function and cataract pathology. Curr Mol Med 2009; 9: 887–892.
Ye M, Berry-Wynne KM, Asai-Coakwell M et al: Mutation of the bone morphogenetic protein GDF3 causes ocular and skeletal anomalies. Hum Mol Genet 2010; 19: 287–298.
Wang L, He F, Bu J et al: ABCB6 mutations cause ocular coloboma. Am J Hum Genet 2012; 90: 40–48.
Helias V, Saison C, Ballif BA et al: ABCB6 is dispensable for erythropoiesis and specifies the new blood group system Langereis. Nat Genet 2012; 44: 170–173.
Chavarria-Soley G, Sticht H, Aklillu E et al: Mutations in CYP1B1 cause primary congenital glaucoma by reduction of either activity or abundance of the enzyme. Hum Mutat 2008; 29: 1147–1153.
Pasutto F, Chavarria-Soley G, Mardin CY et al: Heterozygous loss-of-function variants in CYP1B1 predispose to primary open-angle glaucoma. Invest Ophthalmol Vis Sci 2010; 51: 249–254.
Chambers D, Wilson L, Maden M, Lumsden A : RALDH-independent generation of retinoic acid during vertebrate embryogenesis by CYP1B1. Development 2007; 134: 1369–1383.
Lupo G, Gestri G, O'Brien M et al: Retinoic acid receptor signaling regulates choroid fissure closure through independent mechanisms in the ventral optic cup and periocular mesenchyme. Proc Natl Acad Sci 2011; 108: 8698–8703.
Kloeckener-Gruissem B, Vandekerckhove K, Nurnberg G et al: Mutation of solute carrier SLC16A12 associates with a syndrome combining juvenile cataract with microcornea and renal glucosuria. Am J Hum Genet 2008; 82: 772–779.
Ponnam SP, Ramesha K, Tejwani S, Ramamurthy B, Kannabiran C : Mutation of the gap junction protein alpha 8 (GJA8) gene causes autosomal recessive cataract. J Med Genet 2007; 44: e85.
Burdon KP, Wirth MG, Mackey DA et al: Investigation of crystallin genes in familial cataract, and report of two disease associated mutations. Br J Ophthalmol 2004; 88: 79–83.
Bahn CF, Falls HF, Varley GA, Meyer RF, Edelhauser HF, Bourne WM : Classification of corneal endothelial disorders based on neural crest origin. Ophthalmology 1984; 91: 558–563.
Vincent A, Billingsley G, Priston M et al: Further support of the role of CYP1B1 in patients with Peters anomaly. Mol Vis 2006; 12: 506–510.
Fantes J, Ragge NK, Lynch SA et al: Mutations in SOX2 cause anophthalmia. Nat Genet 2003; 33: 461–463.
Ragge NK, Brown AG, Poloschek CM et al: Heterozygous mutations of OTX2 cause severe ocular malformations. Am J Hum Genet 2005; 76: 1008–1022.
Jimenez NL, Flannick J, Yahyavi M et al: Targeted ‘next-generation’ sequencing in anophthalmia and microphthalmia patients confirms SOX2, OTX2 and FOXE3 mutations. BMC Med Genet 2011; 12: 172.
Acknowledgements
We thank the families for their participation in this research. Support is acknowledged from the NHMRC and the Ophthalmic Research Institute of Australia.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Rights and permissions
About this article
Cite this article
Prokudin, I., Simons, C., Grigg, J. et al. Exome sequencing in developmental eye disease leads to identification of causal variants in GJA8, CRYGC, PAX6 and CYP1B1. Eur J Hum Genet 22, 907–915 (2014). https://doi.org/10.1038/ejhg.2013.268
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2013.268
Keywords
This article is cited by
-
Demographics and histopathological characteristics of enucleated microphthalmic globes
Scientific Reports (2022)
-
BMP3 is a novel locus involved in the causality of ocular coloboma
Human Genetics (2022)
-
The genetic landscape of crystallins in congenital cataract
Orphanet Journal of Rare Diseases (2020)
-
The genetic architecture of aniridia and Gillespie syndrome
Human Genetics (2019)
-
Genetics of anophthalmia and microphthalmia. Part 1: Non-syndromic anophthalmia/microphthalmia
Human Genetics (2019)

{kind=link}
{kind=link}
{kind=link}