
Abstract
The Bcl-2 family of proteins has been characterized by either anti-apoptotic or pro-apoptotic activity. Insight into how Bcl-2 family members function has been gained by determining their intracellular localization. We have generated a monoclonal anti-A1-a antibody and used a COS-7 overexpression system to study the localization of the murine anti-apoptotic Bcl-2 family member, A1-a. A1-a overexpressed in COS-7 cells localized to the nucleus as determined by subcellular fractionation and immunofluorescent microscopy. A1-a in the COS-7 nucleus bound tightly to the nuclear matrix as evidenced by resistance to treatment with DNAse I and RNAse A and sequential extraction with 1.0% Triton X-100, 0.15 M NaCl, 0.25 M HCl, 0.5 M Tris pH 7.4 and 6 M urea. HPLC analysis of A1-a, subsequent to SDS extraction, produced fractions that gave multiple bands when analyzed by Western blot analysis suggesting a propensity to form multimers. COS-7 cells transfected with A1-a were protected from apoptotic induction by staurosporine treatment. Cell Death and Differentiation (2001) 8, 785–793
Similar content being viewed by others


Introduction
The Bcl-2 family of proteins are important mediators of apoptosis. Bcl-2 was the first of the anti-apoptotic proteins to be cloned.1,2 It was initially discovered as a proto-oncogene that did not promote cell proliferation, but instead promoted cell survival.3 While the Bcl-2 related protein CED-9 is the only protein of its kind in C. Elegans,4 mammalian cells have evolved a family of Bcl-2 like proteins with both pro-apoptotic activities, including Bax, Bad, Bid, Bcl-XS and Bak, and anti-apoptotic capacity including Bcl-2, Bcl-XL, Mcl-1, Bcl-w and A1-a (reviewed in5,6,7,8). The logic of evolving multiple proteins within a family may be explained by the expression of these proteins. Numerous studies have shown that whereas multiple Bcl-2 family members may be expressed in a specific cell type and a particular state of cellular differentiation, many of the Bcl-2 family members also differ in their expression pattern allowing for tissue-specific, differentiation-specific and external stimuli-specific expression and regulation.9,10,11,12,13,14,15
In the quest to elucidate the mechanism of action of the Bcl-2 family, numerous studies have focused on determining the subcellular localization of these proteins. Bcl-2 family members can localize to various sites within the cell and different members can be found differentially localized within the same cell type.13,15,16,17,18,19,20,21,22,23,24,25 Localization can be important in Bcl-2 family member function. Mitochondrial Bcl-2 prevents the release of Cytochrome c, thus blocking Cytochrome c-dependent APAF-1 activation of the caspase cascade leading to apoptosis.26,27,28,29 Mutants that abrogate known localization also impede function during apoptosis.22,30,31,32,33 For example, in transiently transfected COS-7 cells, Bcl-2 localizes to the membrane fraction, while Bax is found in the cytosol. Under staurosporine treatment to induce apoptosis, Bax translocates to the mitochondria. Mutations in Bax that block this movement also abrogate apoptosis-promoting activity.23
A1-a is a Bcl-2 family member found primarily in hematopoietic cells.9 Initial studies have shown that A1-a can protect cells from undergoing apoptosis in a manner similar to Bcl-2.34 When 32Dcl3 myeloid precursor cells were induced to undergo apoptosis by growth factor withdrawal, stable transfection with either A1-a or Bcl-2 protected equally against death. Co-transfection with both A1-a and Bcl-2 conferred greater protection than either protein alone, suggesting differences in their mechanism of action.34 When 32Dcl3 cells undergo interleukin 3 withdrawal, a small population undergoes spontaneous neutrophilic differentiation. In the stable transfectants, A1-a was permissive for spontaneous differentiation, while Bcl-2 blocked spontaneous differentiation.
We generated a monoclonal antibody against A1-a in order to determine its subcellular localization. Previous studies in our laboratory have shown that A1-a localizes to the cytoplasm in primary inflammatory macrophages, but can also be found in the nucleus in a fraction of apoptotic cells.35 This study shows that transient overexpression of A1-a in COS-7 cells results solely in nuclear localization as determined by both Western blot analysis and immunofluorescent microscopy. Use of a COS-7 cell overexpression system produced sufficient A1-a to permit biochemical analysis of the extractability of A1-a from the nucleus. While minimal A1-a was extracted with the nuclear membrane fraction, the majority resisted extraction suggesting A1-a could be associated with the nuclear matrix. A1-a was also found to protect COS-7 cells against staurosporine induced apoptosis. These results suggest A1-a is a Bcl-2 family member with a unique localization and that nuclear-localized A1-a is associated with an anti-apoptotic effect.
Results
Generation of A1-a specific monoclonal antibody 20.5
Rats were immunized with partially purified A1-a produced using a prokaryotic expression system and sera were screened using an A1-a ELISA employing A1-a produced with a mammalian expression system. Once a rat with positive serum for anti-A1-a antibody was detected, hybridomas were produced and screened using the mammalian A1-a ELISA. A positive clone, 20.5, was chosen and assayed by Western blot analysis. The monoclonal antibody 20.5 recognizes murine A1-a expressed in transiently transfected COS-7 cells and does not detect proteins in extracts of COS-7 cells transiently transfected with an empty vector (Figure 1). While the COS-7 cell produced A1-a migrates around 17 KD, mass spectrophotometry revealed the protein to be 21 KD as predicted by amino acid sequence analysis (data not shown) suggesting A1-a migrated ahead of the markers and was not digested to a smaller form.

Western blot analysis of A1-a specific antibody. COS-7 cells were transfected with either A1-a or empty vector (mock). Cells were harvested at day 5 after transfection to make whole cell lysates. The lysates were run on a 15% SDS polyacrylamide gel, which was then transferred to a PVDF membrane. The blots were probed initially with sera from A1-a inoculated rats and one positive rat was selected to create a hybridoma culture. Hybridoma clones were screened for anti-A1-a antibody activity and one successful clone was chosen for future experiments. Monoclonal antibody 20.5 recognizes a band in the A1-a transfected COS-7 lysate lane but not in the empty vector (mock) lane showing the specificity to A1-a by the antibody
A1-a localizes to the nucleus of transfected COS-7 cells
Bcl-2 is known to localize to intracellular membranes including mitochondrial, endoplasmic reticular and nuclear membranes.16,17,18,19 To determine if the subcellular distribution of A1-a is similar to Bcl-2, COS-7 cells were transfected with A1-a, harvested by trypsinization, fractionated into nuclear and cytoplasmic fractions and solubilized with Laemmeli buffer 5 days after transfection. The fractions were subjected to Western blot analysis with the A1-a monoclonal antibody. A1-a was found to be exclusively nuclear (Figure 2A). Fractions prepared at days 3, 4, 6 and 7 after transfection also showed exclusively nuclear localization (data not shown). To verify proper subcellular fractionation, the COS-7 A1-a whole cell, nuclear and cytoplasmic fractions were probed for Cytochrome c, an endogenous mitochondria marker. Cytochrome c was found exclusively in the cytoplasmic fraction (Figure 2B) indicating that nuclear localization of A1-a did not result from contamination of the nuclear fraction by cytoplasmic proteins.

A1-a is found in the nuclear fraction of transfected COS-7 cells by Western blot analysis. COS-7 cells were transfected with an A1-a overexpression vector and harvested on days 3, 4, 5, 6 and 7 after transfection. Cells were harvested and made into whole cell lysates. A portion of each lysate was separated into a nuclear and cytoplasmic fraction (see Materials and Methods). The whole cell lysate, nuclear and cytoplasmic fractions were separated on a 15% SDS polyacrylamide gel and subsequently transferred to a PVDF membrane. (A) shows a representative blot probed with the anti A1-a monoclonal antibody, 20.5 from day 5 post transfection. A1-a is found in the whole cell lysate and the nuclear fraction, but not in the cytoplasmic fraction showing A1-a localizes to the nucleus of COS-7 cells. (B) shows a representative blot probed with an anti-cytochrome c antibody. Cytochrome c localizes to the cytoplasmic fraction of the A1-a transfected COS-7 cell
Immunofluorescence microscopy was used to determine if A1-a localized to the nuclear membrane or the entire nucleus. COS-7 cells were grown in chamber slides and 5 days after transfection with A1-a were analyzed using monoclonal antibody 20.5. A1-a was found to localize in the nucleus as detected by confocal microscopy (Figure 3A–D). Cells were counterstained for actin to visualize the cytoplasmic portion of the cell. Cells transfected with the empty vector did not stain with the anti-A1-a antibody (data not shown). As with the Western blot analysis, nuclear A1-a was detectable 1 day after transfection and maximal by 3 days. A1-a in 1 or 2 day transfectants showed either punctate nuclear staining or staining in the nuclear periphery (data not shown). To determine if A1-a localization in the nucleus is an artifact due to overexpression or expression in an atypical cell line, Bcl-2 was also overexpressed in COS-7 cells and examined by confocal microscopy. Bcl-2 in transfected COS-7 cells appears to localize to membranes and mitochondria (Figure 3E–F) which is consistent with previous findings, suggesting that nuclear localization is specific for A1-a in COS-7 cells.

A1-a localizes to the entire nucleus of transfected COS-7 cells. COS-7 cells were transfected with either the A1-a overexpression vector or with Bcl-2. Five days post transfection, cells were harvested to make chamber slides (see Materials and Methods) and fixed in 4% paraformaldehyde. The slides were stained with monoclonal antibody 20.5 to detect A1-a and counterstained with Texas-red Phalloidin which recognizes actin found primarily in the cytoplasm. (A) shows a low power field in phase while (B) shows the A1-a positive cells (green) in the same field. (C) shows A1-a (green) and actin (red), while (D) shows just the actin staining of the same field. Examination by confocal microscopy shows that A1-a is found intranuclearly in COS-7 cells. Empty vector transfected COS-7 cells did not stain for A1-a (data not shown). COS-7 cells transfected with Bcl-2 were stained with a polyclonal antibody against Bcl-2 and counterstained with Texas-red Phalloidin. (E) shows Bcl-2 (green) and actin (red), while (F) shows just the Bcl-2 staining of the same fields. Examination by confocal microscopy shows that Bcl-2 is found in the cytoplasmic portion of the cell consistent with previous findings
A1-a is tightly bound in the nucleus of transfected COS-7 cells
In order to determine the sublocalization of A1-a in the nucleus, the nuclear fraction of A1-a transfected COS-7 cells was subjected to a treatment with DNAse I and RNAse A. This procedure failed to release A1-a from the nuclear fraction (data not shown). The nuclear fraction of A1-a transfected COS-7 cells was then subjected to a sequential extraction protocol. The nuclear pellet was washed twice with 1% Triton-X100, to remove integral membrane proteins,36,37,38 followed by 0.15 M NaCl, which removes peripheral membrane proteins,36,37,38 0.25 M HCl, to remove DNA binding proteins (e.g. histones),39 neutralized with 0.5 M Tris pH 7.4, further washed with 6 M urea to remove nuclear pore proteins36,38 and finally solubilized in a Laemmli buffer containing 2% SDS. The supernatant of each wash and the SDS solubilized pellet were separated using 15% SDS–PAGE and either stained with coomassie blue or analyzed by Western analysis. The coomassie stained gel shows a prominent band co-migrating with A1-a in the SDS solubilized pellet (Figure 4A). It is conceivable that the concentrating effects of the sequential extraction protocol enriched for A1-a sufficiently to visualize an A1-a band by coomassie staining. Western blot analysis confirms the presence of A1-a in the SDS solubilized pellet (Figure 4B) suggesting that A1-a is associated with the nuclear matrix. A small amount of A1-a is present in each of the Triton-X100 wash lanes suggesting some A1-a may also be membrane bound. Lane 7 also shows several bands migrating more slowly than the A1-a monomer. At least some of these bands are present in the whole extract (Figure 2A). They may represent both homo- and heteromultimers of A1-a and their presence indicates that sequential extraction enriches for both monomers and multimers. Slower migrating forms not found in the whole cell extract may be an artifact of the extraction procedure or minor species not appreciated in the whole cell extract.

A1-a remains with the nuclear fraction through a series of extractions. Nuclear fractions were made from COS-7 cells transfected with A1-a. The nuclear fraction was then subjected to a sequential series of extractions. Specifically, nuclei were subjected to two sets of washes with 1% triton-X100 (lanes 1 and 2), followed by 0.15 M NaCl (lane 3), 0.25 M HCl (lane 4), 0.5 M Tris pH 7.4 (lane 5) and 6 M urea (lane 6). The remaining pellet was solubilized in an Laemmli buffer containing 2% SDS (lane 7). Soluble extracts, along with the SDS solubilized pellet were analyzed on a 15% SDS polyacrylamide gel as well as by subsequent Western blot analysis. (A) shows the coomassie blue stained gel of the extracted samples and pellet (molecular weight markers in lane M). The SDS solubilized pellet in lane 7 shows a prominent band migrating similarly to A1-a. (B) shows a Western blot made from a replicate gel and probed with monoclonal antibody 20.5. A1-a is found predominantly in the SDS solubilized pellet
HPLC analysis of nuclear A1-a suggests a tendency for multimerization
After performing the extractions above, the final pellet resuspended in SDS sample buffer was precipitated with acetone and resolubilized in 6 M urea. This sample was analyzed by reversed-phase HPLC and proteins were eluted with increasing concentrations of acetonitrile. All peaks with absorbance at 210 nm were analyzed by Western blot analysis (Figure 5A). Only fractions A, B & C contained detectable A1-a (Figure 5A inset). Western blot analysis from another HPLC separation comparing the HPLC purified fractions with the material loaded onto the column (Figure 5B) shows a major A1-a monomer band and multiple minor bands representing A1-a multimers. The estimated size of the multimers suggests that some of them are A1-a homomultimers. As multimerization remains throughout HPLC separation, this suggests A1-a has a strong proclivity to multimerize. Which of these species are homomultimers or heteromultimers has yet to be determined.

HPLC separation of nuclear extracted A1-a. A1-a transfected nuclei were extracted by a series of sequential reagents as in Figure 4. The SDS solubilized pellet was precipitated with acetone and resuspended in 6 M urea. The A1-a sample was loaded onto a Vydac protein C4 reversed phase column and analyzed by HPLC. (A) shows the elution profile of proteins (absorbance at 210 nm) with increasing concentration of acetonitrile (ACN). (A) inset shows A1-a bands by Western blot analysis corresponding to fractions A, B & C on the chromatogram. (B) shows a Western blot analysis of another HPLC separation comparing the material loaded onto the column (lane 1) with the HPLC purified fractions (lanes 2–4). Subsequent to HPLC separation, A1-a is found in both monomeric and multimeric forms
Nuclear A1-a protects COS-7 cells from staurosporine induced apoptosis
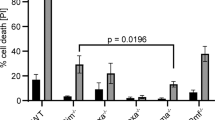
As nuclear A1 has been observed in apoptotic cells,35 both A1-a and empty vector transfected cells were assayed by TUNEL to determine if nuclear A1-a correlated with apoptosis in COS-7 cells. Both A1-a and empty vector transfectants had an equal percentage of TUNEL positive cells, ranging from 1–7%, in three independent experiments. This suggests that A1-a does not induce apoptosis in COS-7 cells. In addition, A1-a transfected COS-7 cells were stained for both A1-a and TUNEL and the few apoptotic cells present were all negative for A1-a (data not shown) suggesting an inverse association between A1-a positive nuclei and apoptosis in this system. In order to determine if nuclear A1-a does protect against apoptosis, COS-7 cells were transfected with either A1-a or empty vector. Three days after transfection, cells were treated with 3 μM staurosporine for up to 6 h. Cells were then harvested each hour for immunofluorescent microscopy and probed for the presence of A1-a as well as apoptosis (Figure 6A–E). Over 300 COS-7 cells were counted on each slide for each timepoint in two separate experiments. COS-7 cells were found to be transfected with 30% efficiency. The amount of apoptosis found at each timepoint reached maximal levels at 4 h (Figure 7). At 4 h, 47% of the mock transfected cells were TUNEL positive, as opposed to 15% among the A1-a transfected cells. The subsequent decrease in per cent TUNEL positivity among the mock transfected cells may be due to loss of cellularity caused by staurosporine-induced death. It is important to note that not a single TUNEL positive cell from the A1-a transfected population was A1-a positive (Figure 7) indicating that A1-a protects COS-7 cells from apoptosis. In addition, staurosporine treatment was extended to 24 and 48 h when almost 100% of untransfected COS-7 cells were killed. At 24 h, the number of A1-a positive cells sharply decreased, even more so at 48 h, yet not a single A1-a positive cell was TUNEL positive at either time point (data not shown). This suggests that A1-a positive cells can die, but only after removal of A1-a.

A1-a transfected COS-7 cells are resistant to staurosporine-induced apoptosis. COS-7 cells were transfected with either A1-a or empty vector. After 3 days of transfection, cells were treated with 3 μM staurosporine for up to 6 h. Cells were stained for A1-a (red) and TUNEL (green). Over 300 COS-7 cells were counted on each slide for each timepoint in two separate experiments. A1-a positive cells were found to be distinct from TUNEL positive cells. (A) and (B) show a representative field of A1-a transfected COS-7 cells in phase and fluorescence, respectively. (C) is the same field as (B), but with only the green fluorescence, showing that none of the A1-a positive cells are TUNEL positive. (D) and (E) show a representative field of empty vector transfected COS-7 cells in phase and fluorescence, respectively

Quantitation of TUNEL positive cells among A1-a or mock vector transfected cells. Three days after transfecting with either A1-a or mock vector, COS-7 cells were treated with staurosporine and harvested hourly for 6 h after treatment. Over 300 cells at each timepoint were assessed for A1-a expression and TUNEL positivity. The per cent TUNEL positive cells from the mock transfected culture (open circles) showed the highest percentage of TUNEL staining. Only the A1-a negative cells from the A1-a transfected culture shown as a percentage of the total culture (closed circles) showed TUNEL staining. A1-a expressing cells from the A1-a transfected culture were always TUNEL negative (closed squares)
Discussion
We have shown that murine A1-a can localize within the nucleus of COS-7 cells upon transient transfection with an A1-a expression vector. Previous studies in our laboratory have shown that nuclear A1-a occurs in some inflammatory leukocytes undergoing apoptosis.35 By using a COS-7 overexpression system, we were able to examine nuclear localization more closely. By both biochemical and immunofluorescent analysis, A1-a was found to be exclusively nuclear, in contrast to our in vivo results which showed predominantly cytoplasmic localization.35 Recently, two studies have shown that Bcl-2, Bax and Mcl-1 can display a mixed pattern of cytoplasmic and intranuclear localization.24,25 In contrast, we show that A1-a can adopt an exclusively intranuclear localization, even in a cell type in which Bcl-2 is restricted to the cytoplasm. A1-a does not have a known nuclear localization sequence. However, other nuclear proteins without nuclear localization sequences are known to be shuttled to the nucleus by escort proteins.40
A1-a overexpressed in COS-7 cells appeared to distribute to the nuclear matrix. Extraction procedures that removed membrane proteins, pore proteins and DNA associated proteins did not extract A1-a, suggesting localization to the nuclear matrix. It has been shown that the viral Bcl-2 family member, E1B 19K, binds to lamins, which are found in the nuclear lamina underlying the inner nuclear membrane. Mutations that cause mislocalization of the lamins, and thus change the localization of E1B 19K protein, lead to abrogation of the E1B 19K anti-apoptotic activity.41 A possible mechanism E1B 19K may use to block apoptosis may involve sequestration of pro-apoptotic proteins. The E1B 19K protein has been shown to sequester CED-4, the C. elegans APAF-1 homologue, from the cytosol to the nucleus, abrogating the ability of CED-4 to activate caspase-8.42 The E1B 19K protein also blocks apoptotic induction by sequestering FADD, a mammalian caspase recruiting protein that functions in Fas mediated apoptosis.43 E1B 19K can also confer protection by binding and sequestering Btf, a transcription factor that induces apoptosis upon overexpression.44 Given the difficulty in extracting A1-a from the COS-7 nucleus and its tendency to multimerize, it would be easy to believe that A1-a might function in an anti-apoptotic manner via protein-protein interaction. Further studies are needed to determine which proteins A1-a interacts with and if A1-a sequesters these proteins during apoptotic induction.
The ability to regulate transcription could be another possible mechanism in which A1-a could exert a protective effect. It has already been shown that Bcl-2 family members can regulate transcription. It is unclear if the Bcl-2-like proteins interact directly or indirectly with transcription factors, but in some cases, this form of association is associated with protection against apoptosis.45,46,47,48,49,50,51 The nuclear localization of A1-a would make the targeting of a transcription factor much easier and possibly through direct interaction. All together, these results show that different Bcl-2 family proteins can share and diverge in their methods of blocking apoptosis and that the difference in localization between A1-a and Bcl-2 may lead to differing mechanisms of protection. This is also suggested by data from our lab showing that A1-a and Bcl-2 co-transfectants have a greater survival effect than each protein alone in factor deprived 32Dcl3 cells.34
In addition to the novel localization pattern of A1-a, we have also shown that A1-a expressing COS-7 cells are not apoptotic following exposure to staurosporine. While long term exposure (24–48 h) to staurosporine caused most cells to die, only A1-a negative cells were found to die at any time after staurosporine treatment. Even though the percentage of A1-a positive cells declined over this treatment period, the A1-a positive cells did remain viable as determined by TUNEL staining. While previous findings from our lab have shown that only apoptotic cells could occasionally contain nuclear A1-a,35 our results are not inconsistent with the previous finding. Previously, we have shown that A1-a was found in the nucleus of cells only when that cell was apoptotic. Those cells were activated inflammatory cells and could have multiple pathways for apoptotic induction, including one that is A1-a independent. The question remains how does A1-a exert its function from the nucleus. Since staurosporine is a protein kinase C inhibitor, it acts to induce apoptosis in the cytoplasm. If A1-a acts to sequester factors as some of its family members are known to do, it could easily sequester vital pro-apoptotic components from the cytoplasm to the nucleus, away from the staurosporine induced pathway.
Materials and Methods
Monoclonal antibody generation
Female LEW/CrlBr rats were immunized by i.p. injection of 100 μg of partially purified A1-a produced from a prokaryotic expression system.34 Prior to injection, A1-a was conjugated to KLH (Pierce) and diluted in an equal volume of adjuvant (Ribi). Bleeds were examined for anti-A1-a activity by ELISA (see below). Rats with positive sera were given 100 μg of A1-a i.p. 3 days prior to sacrifice. At sacrifice, the spleen was removed and the hematopoietic cells isolated were fused with Ag8.653 myeloma cells52 in a 4 : 1 ratio with polyethylene glycol as described before.53 Successful fusions were selected using HAT medium. These cells were then cloned in soft agarose containing HAT medium at a concentration of 2000 cells/ml/60 mm plate. Clones were picked and grown to generate supernatant that was assayed for anti-A1-a antibody activity by ELISA.
Transfection and harvest of COS-7 cells
COS-7 cells were transfected by the DEAE-dextran method as described before.54 Briefly, 20 h before transfection, cells were plated on 100 mm tissue culture dishes (Corning) at 1.8×106 cells/dish in 10 ml of complete medium. After 20 h, the medium was aspirated and the cells washed once with 5 ml of PBS. Four milliliters of DMEM supplemented with 10% Nu-serum IV (Collaborative Research) was added to the plate. Ten μg of plasmid DNA containing the SV40 virus replication origin (either parent vector RJB10B54 or RJB-A1-a34 or pRC/CMV-Bcl255) in 0.2 ml of tris-buffered saline at pH 7.4 was mixed with 0.8 ml of serum free DMEM containing 1 mg/ml DEAE-dextran and 250 μM chloroquine and incubated with COS-7 cells at 37°C for 4 h.
The transfection solution was then aspirated and incubated with 5 ml of 10% DMSO in PBS for 4 min. After removing the DMSO, cells were washed once in 5 ml of PBS and incubated in 10 ml of complete medium until harvest. Five days post transfection, cells were harvested in one of three ways. First, COS-7 cell lysates were made by trypsinizing the cells from the plates, pelleting the cells by centrifugation and solubilizing the pellet in a modified Laemmli gel sample buffer.34 Second, slides were made by lifting transfected cells from the tissue culture plate by trypsinization and seeding chamber slides (Lab-Tek) with 105 cells per chamber. The slides were then incubated at 37°C overnight, fixed in cold paraformaldehyde (4% in PBS) for 15 min, washed three times in PBS and stored at 4°C in PBS with 0.002% azide. Third, COS-7 cells were trypsinized and pelleted by centrifugation as above. Pellets were resuspended in a hypotonic solution of 10 μM HEPES, 15 μM KCl, 2 μM MgCl2, 0.1 μM EDTA, 1 μM DTT and the protease inhibitors PMSF, leupeptin and pepstatin and placed on ice for 10 min. Cells were disrupted by using a dounce homogenizer (Radnoti) until microscopic analysis showed >95% free nuclei or disrupted cells. The nuclei were pelleted by centrifugation at 800×g for 5 min; the supernatant was mixed with a 2X Laemmli buffer and saved as the cytoplasmic fraction. The pellet was resuspended in the hypotonic solution and NP-40 (Sigma) was added to a final concentration of 0.1%. This was vortexed and pelleted as before. The supernatant was removed and the pellet was solubilized in Laemmli buffer.
ELISA
Lysates from A1-a transfected and mock vector transfected COS-7 cells were dissolved in 2–4 volumes of guanidinium HCl and diluted in ELISA Coating Buffer (0.1 M NaHCO3 in PBS, pH 8.2) to a final concentration of 10 μg/ml. ELISA plate wells were coated with 100 μl of this solution and incubated at 4°C overnight. The wells were washed three times with washing buffer (0.05% Tween-20 in PBS) and then coated with 200 μl of 3% BSA (Sigma) in PBS and incubated at 4°C overnight. The wells were then washed twice as before and then 100 μl of either rat serum or hybridoma supernatant was added and incubated at 37°C for 1 h. After two more rounds of washing, 100 μl of a goat anti-rat antibody conjugated to HRP (Santa Cruz) was added at 1 : 1000 dilution to each well and incubated at 37°C for 1 h. Each well was then washed four times and 50 μl of TMB solution (1 mg TMB (Sigma) dissolved in 1 ml DMSO plus 9 ml 0.1 M sodium acetate pH 6 and 3.3 μl of H2O2) were added to each well. Wells were incubated at room temperature for 30 min under cover and then 50 μl of 1 M H2SO4 was added to stop the reaction. A1-a positive sera or supernatants were determined by measuring absorbance at 450 nm.
Western blot analysis
COS-7 cell lysates were analyzed for protein by the Bradford assay (Bio-Rad). Samples were separated on 15% SDS–PAGE gels and transferred to Immobilon-P membranes (Millipore) by wet electrophoresis transfer (Bio-Rad) in 25 mM Tris, 190 mM glycine for 40 min at 100 V. The membranes were blocked with 5% dry milk in PBS (block solution) and probed with monoclonal antibody 20.5 hybridoma supernatant or anti-Cytochrome c monoclonal antibody (Pharmingen) at 1 : 1000 dilution overnight at 4°C. The blot was then washed three times in PBS containing 0.5% Tween-20 for 15 min per wash, blocked again for 20 min, incubated with either goat anti-rat or goat anti-mouse antibody conjugated to HRP (Santa Cruz) at 1 : 2000 and then washed again as before. Detection was by enhanced chemiluminescence following the supplier's recommended protocol (Amersham).
Extraction of COS-7 cell nuclei
Nuclease facilitated extraction was performed as described before.56 Briefly, COS-7 nuclei were resuspended in 0.1 mM MgCl2 and diluted fivefold with 4 volumes of 10% (w/v) sucrose, 10 mM TEA-HCl (pH 8.5), 0.1 mM MgCl2, 0.5 mM PMSF and 1 mM DTT. Five μg of DNAse I and 1 μg RNAse A were added per milliliter, and the sample was incubated for 15 min at room temperature. After underlayering with one volume of ice cold 30% sucrose, 10 mM TEA-HCl (pH 7.4), 1 mM MgCl2, 0.5 mM PMSF and 1 mM DTT, the sample was pelleted by centrifugation at 20 000×g. The supernatant was collected and the pellet resuspended by vortexing in 10% sucrose, 10 mM TEA-HCl (pH 7.4), 0.1 mM MgCl2, 0.5 mM PMSF and 1 mM DTT. Twenty-five μg of DNAse I and 5 μg of RNAse A were then added per milliliter. The sample was incubated and pelleted as before except that the underlayering solution was 30% sucrose, 10 mM TEA-HCl (pH 7.4), 5 mM MgCl2 0.5 mM PMSF and 1 mM DTT. The supernatant was collected and the pellet resuspended in a Laemmli buffer.
Sequential extraction of A1-a transfected COS-7 cell nuclei was performed in the following manner. The nuclear pellet, produced from homogenization and hypotonic washing, was resuspended in 1 ml of a 1% Triton-X100 solution, vortexed vigorously and then pelleted by centrifugation for 10 min at 4°C. The supernatant was collected and the pellet washed in a further 1 ml of 1% Triton-X100. The suspension was vortexed, pelleted and collected as before. The pellet was then washed sequentially with 1 ml solutions of the following: 0.15 M NaCl placed on ice for 5 min, 0.25 M HCl placed on ice for 5 min, 0.5 M Tris pH 7.4, 6 M urea and finally a Laemmli buffer containing 2% SDS. Supernatants from each wash were stored at −20°C. For Western blot analysis, 15 μl of each supernatant was mixed with 15 μl of a 2X SDS Laemmli buffer, boiled for 5 min and then loaded on a gel for analysis. 15 μl of the SDS Laemmli buffer containing the final pellet was used undiluted.
Immunofluorescent microscopy
COS-7 cells seeded in chamber slides were harvested as described above and permeabilized in 0.2% Triton-X100 in PBS for 15 min. Cells were rinsed in PBS three times and then blocked in PBS containing 0.05% Triton-X100, 2% BSA and 1% goat serum for 30 min. The cells were then probed with anti-A1-a monoclonal antibody 20.5 culture supernatant undiluted or anti-Bcl-2 monoclonal clone 4D7 at 1 : 100 (Pharmingen) for 3 h. Cells were washed three times with 0.05% Triton-X100 in PBS for 15 min per wash. Cells were blocked again for another 30 min and then probed with either FITC-goat anti-rat IgG or FITC-goat anti-rabbit IgG (Santa Cruz) for 1 h. The cells were then washed several times in 0.05% Triton-X100 in PBS for 3 min per wash. Following a further wash in PBS, cells were counterstained for actin with Texas-Red phalloidin (Molecular Probes) for 1.5 h according to the manufacturer's protocol. The cells were washed three times in PBS for 5 min per wash and then mounted using the ProLong Antifade kit (Molecular Probes). When TUNEL staining was performed, it was done prior to probing with the A1-a monoclonal antibody. TUNEL staining assay was performed as per manufacturer's protocol (Boehringer Mannheim). Slides were examined by confocal microscopy. Images were captured using both phase and fluorescent microscopy including both FITC and Texas-Red filters. Images from each filter were overlaid to form the composite figure or presented separately as in Figure 6C.
HPLC
The nuclear fraction of A1-a transfected COS-7 cells was subjected to the sequential extraction protocol described above. 250 μl of the final extract in Laemmli buffer containing 2% SDS was removed and 1 ml of cold acetone was added. This was vortexed and stored overnight at −20°C. The resulting precipitate was pelleted by centrifugation at 16 000×g at 4°C for 15 min. The supernatant was removed and the pellet was washed with 1 ml of cold acetone overnight followed by centrifugation. The pellet was quickly dried using vacuum centrifugation and then dissolved in 6 M urea. A1-a in 6 M urea was loaded onto a Vydac protein C4 reversed-phase column (250×4.6 mm, Vydac). The gel was equilibrated with 0.1% (v/v) trifluoroacetic acid in 90% water and 10% acetonitrile. A1-a was eluted with a two-step linear gradient to 80% acetonitrile in 1% aqueous trifluoroacetic acid at a flow rate of 1 ml/min.
Abbreviations
- TUNEL:
-
dUTP nick end labeling terminal transferase
- PVDF:
-
poly (vinylidene difluoride)
- PMSF:
-
phenyl-methyl-sulphonyl-fluoride
References
Tsujimoto Y, Finger LR, Yunis J, Nowell PC, Croce CM . 1984 Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation Science 226: 1097–1099
Bakhshi A, Jensen JP, Goldman P, Wright JJ, McBride OW, Epstein AL, Korsmeyer SJ . 1985 Cloning the chromosomal breakpoint of t(14;18) human lymphomas: clustering around JH on chromosome 14 and near a transcriptional unit on 18 Cell 41: 899–906
Vaux DL, Cory S, Adams JM . 1988 Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells Nature 335: 440–442
Hengartner MO, Horvitz HR . 1994 C. Elegans cell survival gene ced-9 encodes a functional homolog of the mammalian proto-oncogene bcl-2 Cell 76: 665–676
Brown R . 1996 The Bcl-2 family of proteins Br. Med. Bull. 53: 466–477
Reed JC . 1998 Bcl-2 family proteins Oncogene 17: 3225–3236
Minn AJ, Swain RE, Ma A, Thompson CB . 1998 Recent progress on the regulation of apoptosis by Bcl-2 family members Adv. Immunol. 70: 245–279
Newton K, Strasser A . 1998 The Bcl-2 family and cell death regulation Curr. Opin. Genet. Dev. 8: 68–75
Lin EY, Orlofsky A, Berger MS, Prystowsky MB . 1993 Characterization of A1, a novel hematopoietic-specific early-response gene with sequence similarity to Bcl-2 J. Immunol. 151: 1979–1988
Krajewski S, Krajewski M, Shabaik A, Wang HG, Irie S, Fong L, Reed JC . 1994 Immunohistochemical analysis of in vivo patterns of Bcl-X expression Cancer Res. 54: 5501–5507
Gonzalez-Garcia M, Garcia I, Ding L, O'Shea S, Boise LH, Thompson CB, Nuñez G . 1995 Bcl-x is expressed in embryonic and postnatal neural tissues and functions to prevent neuronal cell death Proc. Natl. Acad. Sci. 92: 4304–4308
Boise LH, Minn AJ, Noel PJ, June CH, Accavitti MA, Lindsten T, Thompson CB . 1995 CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-Xl Immunity 3: 87–98
Yang T, Kozopas KM, Craig RW . 1995 The intracellular distribution and pattern of expression of Mcl-1 overlap with, but are not identical to, those of Bcl-2 J. Cell Biol. 128: 1173–1184
Yang T, Buchan HL, Townsend KJ, Craig RW . 1996 Mcl-1, a member of the Bcl-2 family, is induced rapidly in response to signals for cell differentiation or death, but not to signals for cell proliferation J. Cell. Physiol. 166: 523–536
Krajewski S, Krajewski M, Reed JC . 1996 Immunohistochemical analysis of in vitro patterns of bak expression, a proapoptotic member of the Bcl-2 protein family Cancer Res. 56: 2849–2855
Hockenberry D, Nuñez G, Milliman C, Schreiber RD, Korsmeyer SJ . 1990 Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death Nature 348: 334–336
Monaghan P, Robertson D, Amos TA, Dyer MJ, Mason DY, Greaves MF . 1992 Ultrastructural localization of bcl-2 protein J. Histochem. Cytochem. 40: 1819–1825
Krajewski S, Tanaka S, Takayama S, Schibler MJ, Fenton W, Reed JC . 1993 Investigation of the subcellular distribution of the Bcl-2 oncoprotein: residence in the nuclear envelope, endoplasmic reticulum, and outer mitochondrial membranes Cancer Res. 53: 4701–4714
Akao Y, Otsuki Y, Kataoka S, Ito Y, Tsujimoto Y . 1994 Multiple subcellular localization of bcl-2: detection in nuclear outer membrane, endoplasmic reticulum membrane, and mitochondrial membranes Cancer Res. 54: 2468–2471
Gonzalez-Garcia M, Perez-Ballestero R, Ding L, Duan L, Boise LH, Thompson CB, Nuñez G . 1994 Bcl-XL is the major Bcl-x mRNA form expressed during murine development and its product localizes to mitochondria Development 120: 3033–3042
Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ . 1996 Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L) Cell 87: 619–628
Hsu YT, Wolter KG, Youle RJ . 1997 Cytosol-to-membrane redistribution of Bax and Bcl-Xl during apoptosis Proc. Natl. Acad. Sci. 94: 3668–3672
Wolter KG, Hsu YT, Smith CL, Nechushtan A, Xi XG, Youle RJ . 1997 Movement of Bax from the cytosol to mitochondria during apoptosis J. Cell Biol. 139: 1281–1292
Hoetelmans RWM, van Slooten HJ, Keijzer R, Erkeland S, van de Velde CJH, van Dierendonck JH . 2000 Bcl-2 and Bax proteins are present in interphase nuclei of mammalian cells Cell Death and Differentiation 7: 384–392
Leuenroth SJ, Grutkoski PS, Ayala A, Simms HH . 2000 The loss of Mcl-1 expression in human polymorphonuclear leukocytes promotes apoptosis J. Leukoc. Biol. 68: 158–166
Zou H, Henzel WJ, Liu X, Lutschg A, Wang X . 1997 Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3 Cell 90: 405–413
Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X . 1997 Cytochrome C and dATP-dependent formation of APAF-1/caspase-9 complex initiates an apoptotic protease cascade Cell 91: 479–489
Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD . 1997 The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis Science 275: 1132–1136
Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, Jones DP, Wang X . 1997 Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked Science 275: 1129–1132
Nguyen M, Branton PE, Walton PA, Oltvai ZN, Korsmeyer SJ, Shore GC . 1994 Role of membrane anchor domain of Bcl-2 in suppression of apoptosis caused by E1B-defective adenovirus J. Biol. Chem. 269: 16521–16524
Zhu W, Cowie A, Wasfy GW, Penn LZ, Leber B, Andrews DW . 1996 Bcl-2 mutants with restricted subcellular location reveal spatially distinct pathways for apoptosis in different cell types EMBO J. 15: 4130–4141
Lee ST, Hoeflich KP, Wasfy GW, Woodgett JR, Leber B, Andrews DW, Hedley DW, Penn LZ . 1999 Bcl-2 targeted to the endoplasmic reticulum can inhibit apoptosis induced by Myc but not etoposide in Rat-1 fibroblasts Oncogene 18: 3520–3528
Nouraini S, Six E, Matsuyama S, Krajewski S, Reed JC . 2000 The putative pore-forming domain of Bax regulates mitochondrial localization and interaction with Bcl-X(L) Mol. Cell. Biol. 20: 1604–1615
Lin EY, Orlofsky A, Wang HG, Reed JC, Prystowsky MB . 1996 A1, a bcl-2 family member, prolongs cell survival and permits myeloid differentiation Blood 87: 983–992
Orlofsky A, Somogyi RD, Weiss LM, Prystowsky MB . 1999 The murine antiapoptotic protein A1 is induced in inflammatory macrophages and constitutively expressed in neutrophils J. Immunol. 163: 412–419
Shen WC, Selvakumar D, Stanford DR, Hopper A . 1993 The sacharomyces cerevisiae LOS1 gene involved in pre-tRNA splicing encodes a nuclear protein that behaves as a component of the nuclear matrix J. Biol. Chem. 268: 19,436–19,444
Aris JP, Blobel G . 1988 Identification and characterization of a yeast nucleolar protein that is similar to a rat liver nucleolar protein J. Cell Biol. 107: 17–31
Pandey S, Parnaik VK . 1991 Identification and characterization of nuclear location signal-binding proteins in nuclear envelopes Biochim. Biophys. Acta 1063: 81–89
Busch H . 1968 Isolation and purification of nuclear proteins Methods Enzymol. 12: 65–84
Turpin P, Hay RT, Dargemont C . 1999 Characterization of IκBα nuclear import pathway J. Biol. Chem. 274: 6804–6812
Rao L, Modha D, White E . 1997 The E1B 19K protein associates with lamins in vivo and its proper localization is required for inhibition of apoptosis Oncogene 15: 1587–1597
Han J, Wallen HD, Nuñez G, White E . 1998 E1B 19,000-molecular-weight protein interacts with and inhibits CED-4-dependant, FLICE-mediated apoptosis Mol. Cell. Biol. 18: 6052–6062
Perez D, White E . 1998 E1B 19K inhibits Fas mediated apoptosis through FADD-dependent sequestration of FLICE J. Cell Biol. 141: 1255–1266
Kasof GM, Goyal L, White E . 1999 Btf, a novel death-promoting transcriptional repressor that interacts with Bcl-2-related proteins Mol. Cell. Biol. 19: 4390–4404
Shen Y, Shenk T . 1994 Relief of p53-mediated transcriptional repression by the adenovirus E1B 19-kDa protein or the cellular Bcl-2 protein Proc. Natl. Acad. Sci. 91: 8940–8944
Sabbatini P, Chiou SK, Rao L, White E . 1995 Modulation of p53-mediated transcriptional repression and apoptosis by the adenovirus E1B 19K protein Mol. Cell. Biol. 15: 1060–1070
Linette GP, Li Y, Roth K, Korsmeyer SJ . 1996 Cross talk between cell death and cell cycle progression: Bcl-2 regulates NFAT-mediated activation Proc. Natl. Acad. Sci. 93: 9545–9552
Grimm S, Bauer MKA, Baeuerle PA, Schulze-Osthoff K . 1996 Bcl-2 down regulates the activity of transcription factor NF-κB induced upon apoptosis J. Cell Biol. 134: 13–23
Miyashita T, U M, Inoue T, Reed JC, Yamada M . 1997 Bcl-2 relieves the trans-repressive function of the glucocorticoid receptor and inhibits the activation of CPP32-like cysteine proteases Biochem. Biophys. Res. Commun. 233: 781–787
Shibasaki F, Kondo E, Akagi T, McKeon F . 1997 Suppression of signaling through transcription factor NF-AT by interactions between calcineurin and Bcl-2 Nature 386: 728–731
See RH, Shi Y . 1998 Adenovirus E1B 19,000-molecular-eight protein activates c-Jun N-terminal kinase and c-Jun mediated transcription Mol. Cell. Biol. 18: 4012–4022
Kearney JF, Radbruch A, Liesegang B, Rajewsky K . 1979 A new mouse myeloma cell line that has lost immunoglobin expression but permits the construction of antibody-secreting hybrid cell lines J. Immunol. 123: 1548–1550
Fazekas de St. Groth S, Scheidegger D . 1980 Production of monoclonal antibodies: strategy and tactics J. Immunol. Methods. 35: 1–21
Altmann SW, Johnson GD, Prystowsky MB . 1991 Single proline substitutions in predicted alpha-helices of murine granulocyte-macrophage colony-stimulating factor result in a loss in bioactivity and altered glycosylation J. Biol. Chem. 266: 5333–5341
Froesch BA, Aime-Sempe C, Leber B, Andrews D, Reed JC . 1999 Inhibition of p53 transcriptional activity by Bcl-2 requires its membrane-anchoring domain J. Biol. Chem. 274: 6469–6475
Davis LI, Blobel G . 1986 Identification and characterization of a nuclear pore complex protein Cell 45: 699–709
Acknowledgements
This work was supported by funds from National Institutes of Health Grant AI-43401 (to MB Prystowsky). We thank Dr. John C Reed for the generous gift of the pRC/CMV-Bcl-2 plasmid and we thank Dr. Matthew D Scharff, Susan Buhl and the Hybridoma Facility of the Cancer Center of the Albert Einstein College of Medicine (CA 13330) for helping to generate the monoclonal antibody. We also thank Michael Cammer and the staff of the Analytical Imaging Facility the Albert Einstein College of Medicine for gracious help in the confocal microscopy.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by JM Hardwick
Rights and permissions
About this article
Cite this article
Somogyi, R., Wu, Y., Orlofsky, A. et al. Transient expression of the Bcl-2 family member, A1-a, results in nuclear localization and resistance to staurosporine-induced apoptosis. Cell Death Differ 8, 785–793 (2001). https://doi.org/10.1038/sj.cdd.4400879
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4400879
Keywords
This article is cited by
-
Bfl-1/A1 functions, similar to Mcl-1, as a selective tBid and Bak antagonist
Oncogene (2008)
-
Constitutive proteasome-mediated turnover of Bfl-1/A1 and its processing in response to TNF receptor activation in FL5.12 pro-B cells convert it into a prodeath factor
Cell Death & Differentiation (2005)
-
Bfl-1S, a novel alternative splice variant of Bfl-1, localizes in the nucleus via its C-terminus and prevents cell death
Oncogene (2003)
