Maintaining Genome Integrity during Seed Development in Phaseolus vulgaris L.: Evidence from a Transcriptomic Profiling Study

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. RNA Extraction, Quantification, and Quality Assessment
2.3. Massive Analysis of 3’-cDNA Ends and Data Analysis
2.4. Primers and Probe Design
2.5. cDNA Synthesis and QuantStudio™ 3D Digital PCR
2.6. Bioinformatic Analysis
3. Results
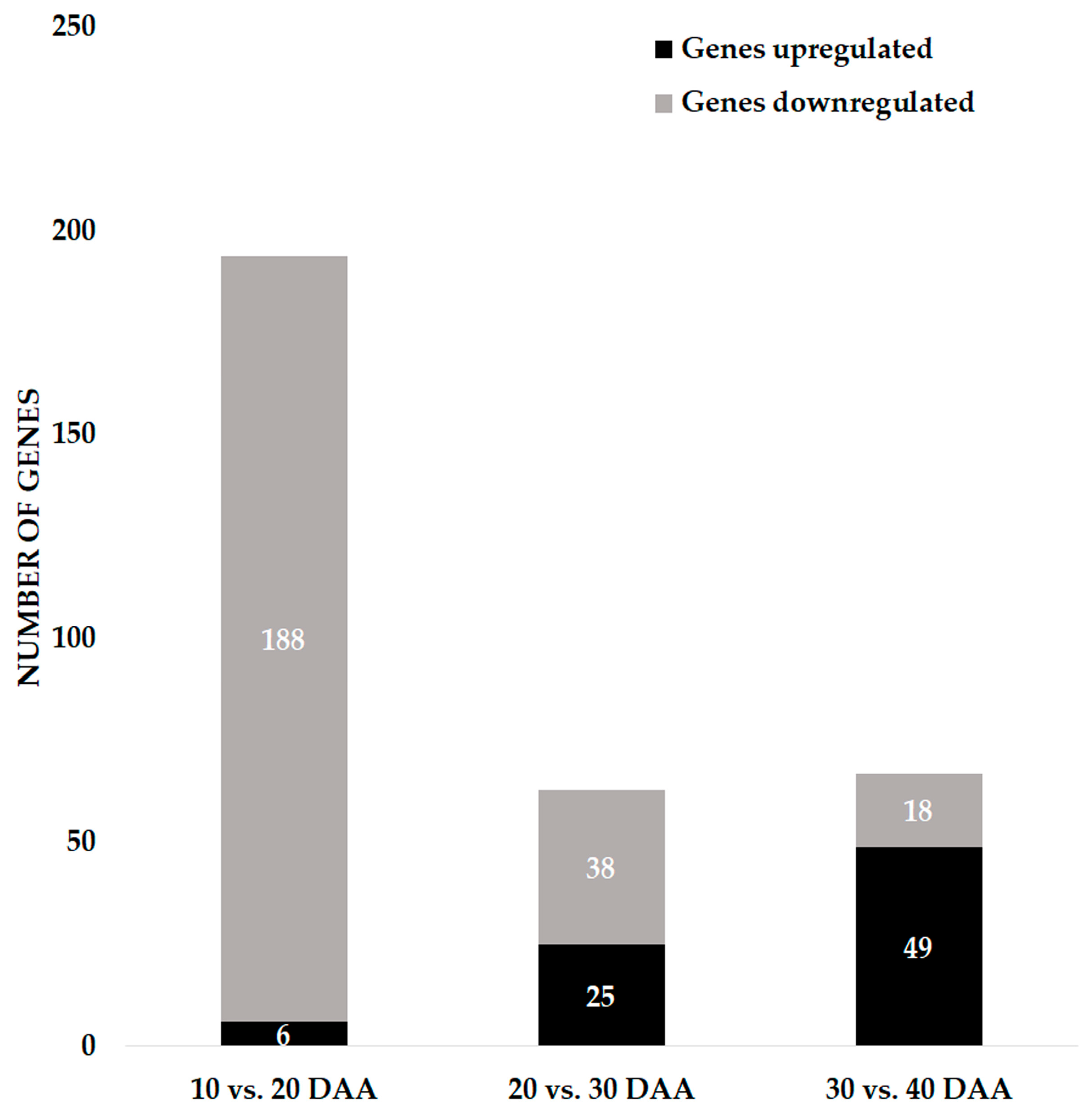
3.1. Global Overview of the Gene Expression during Seed Development
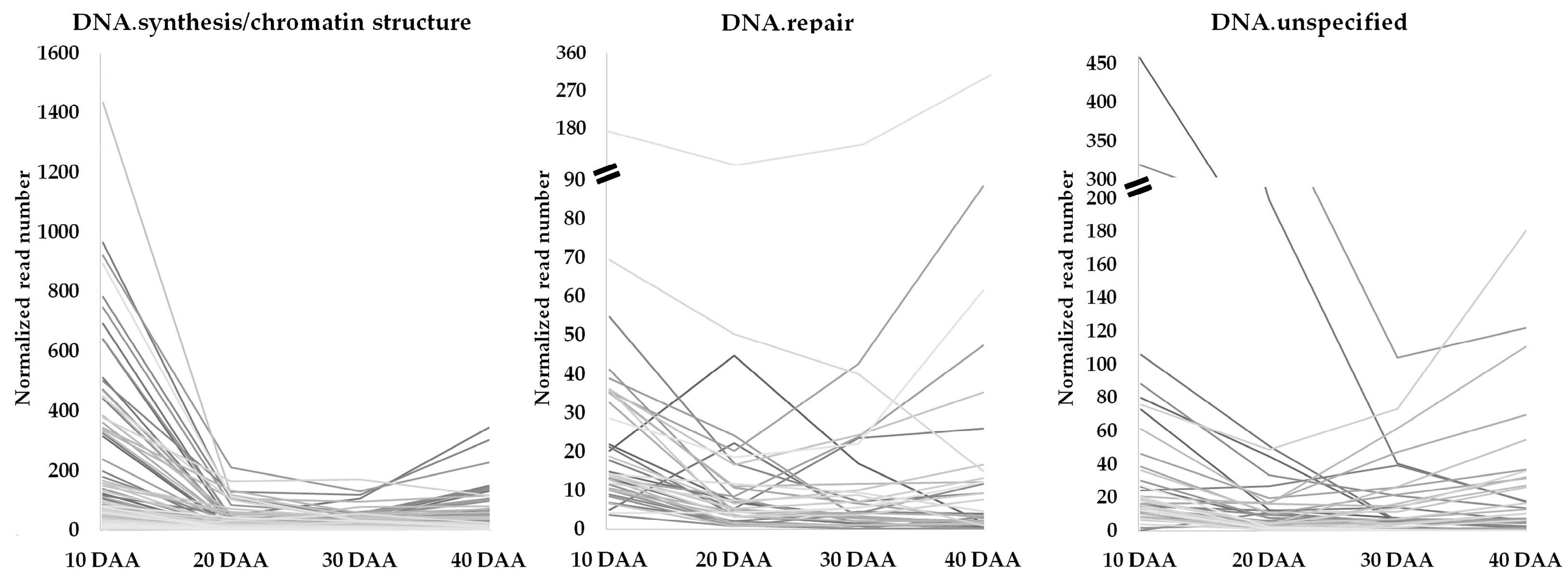
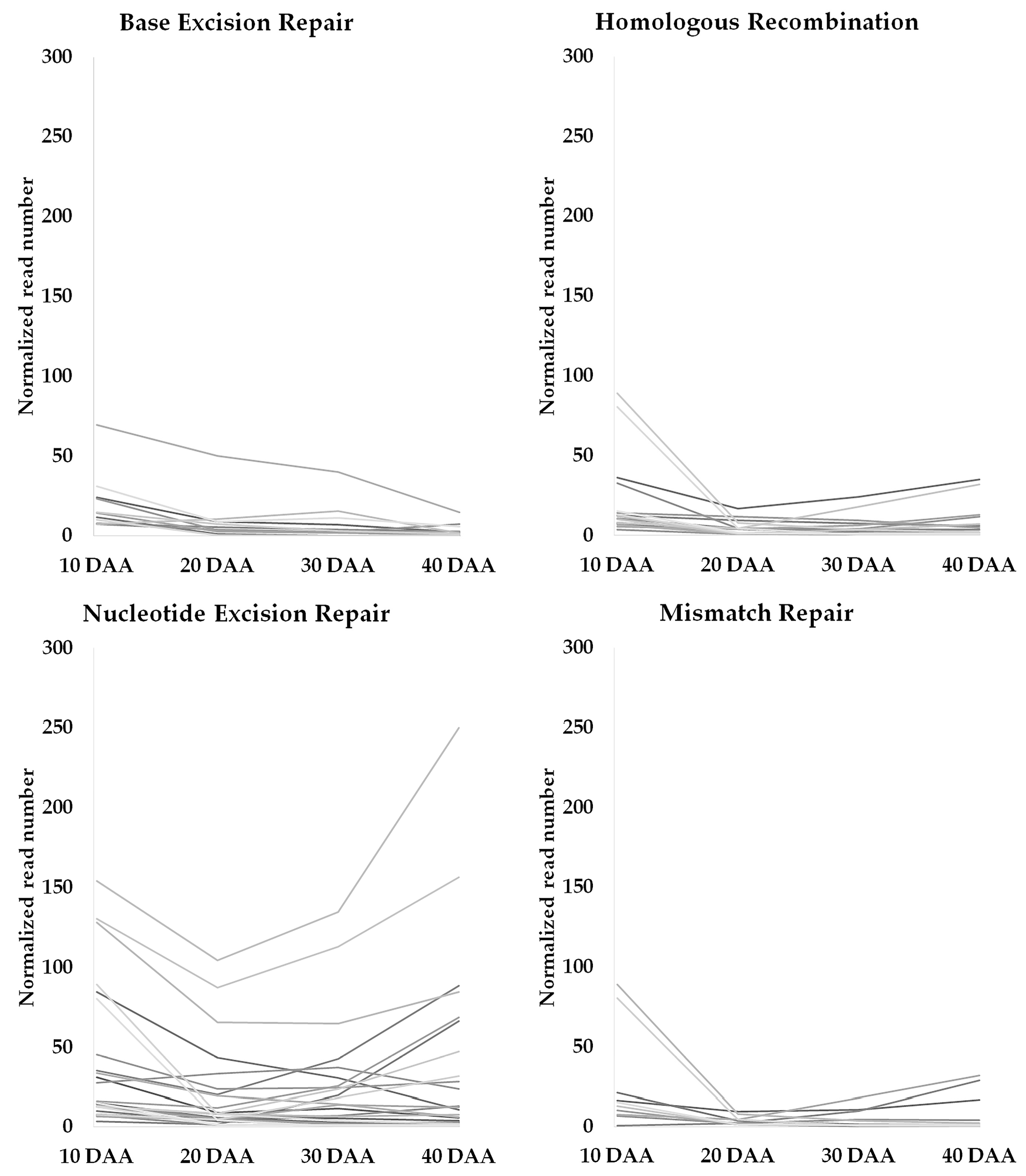
3.2. Genes Involved in DNA Damage Response and Chromatin Remodeling during Seed Development
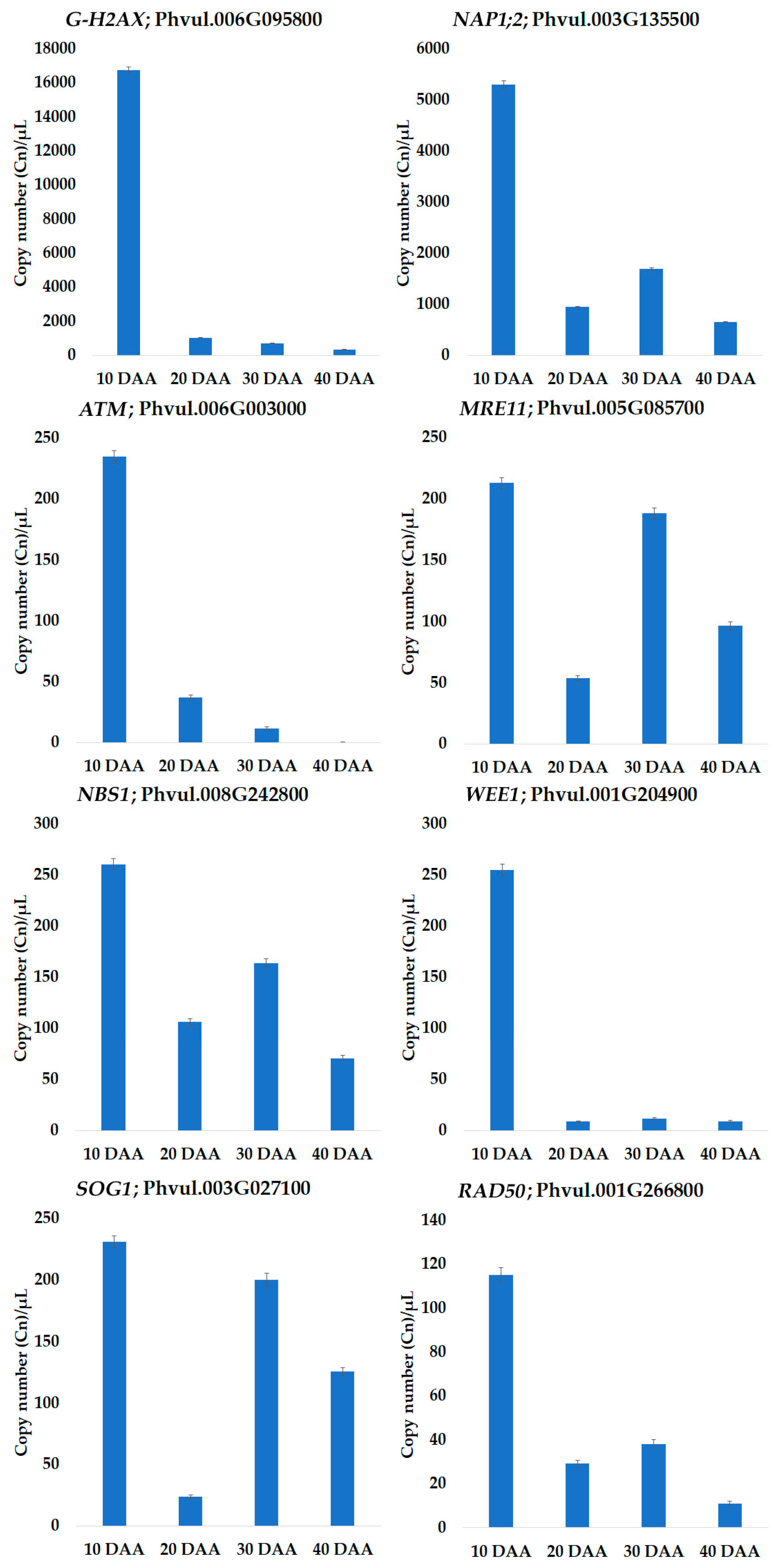
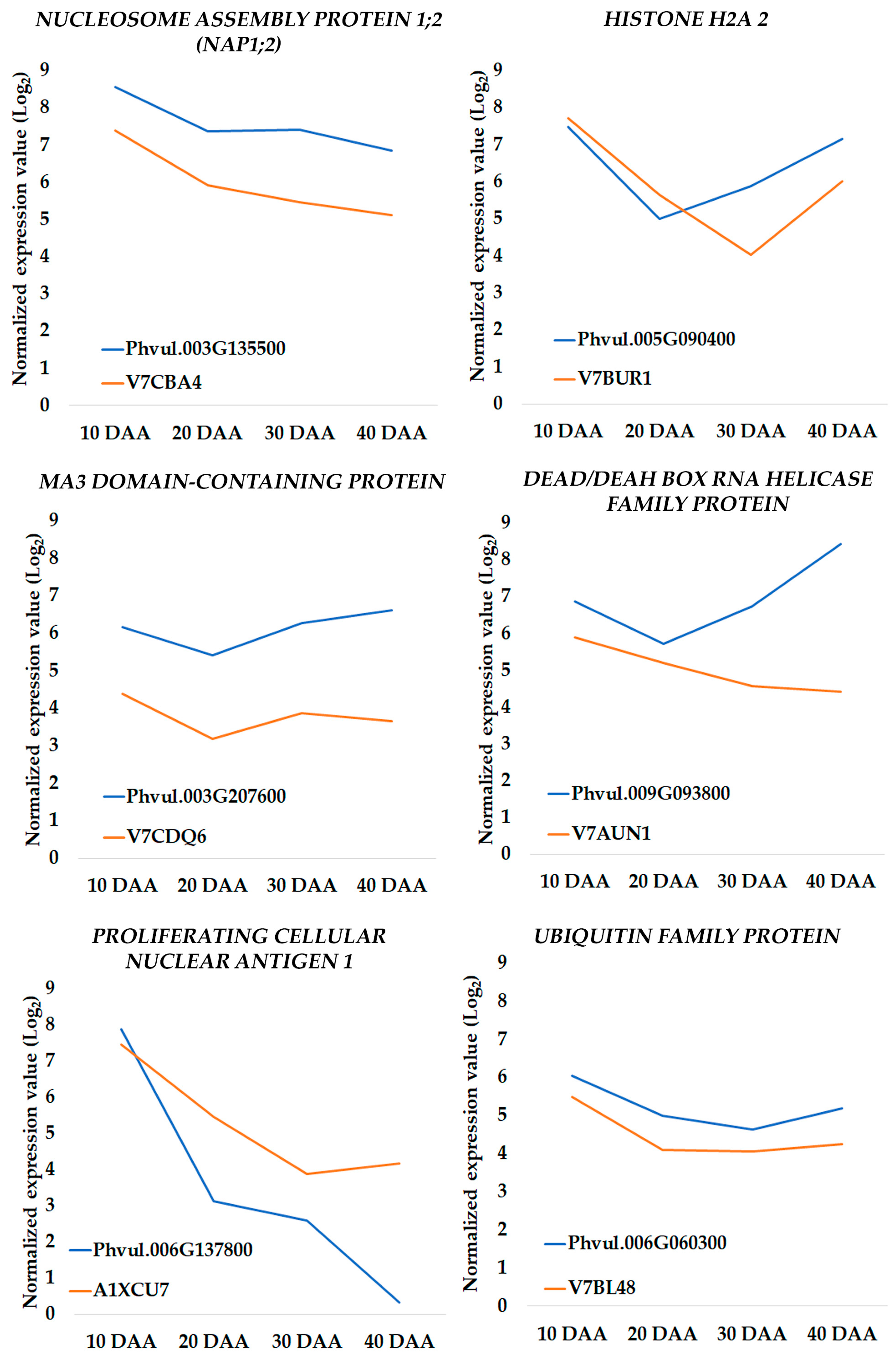
3.3. Quantification by QuantStudio™ 3D Digital PCR
3.4. Changes in Transcriptomic Profiles Are in Accordance with Proteome Changes
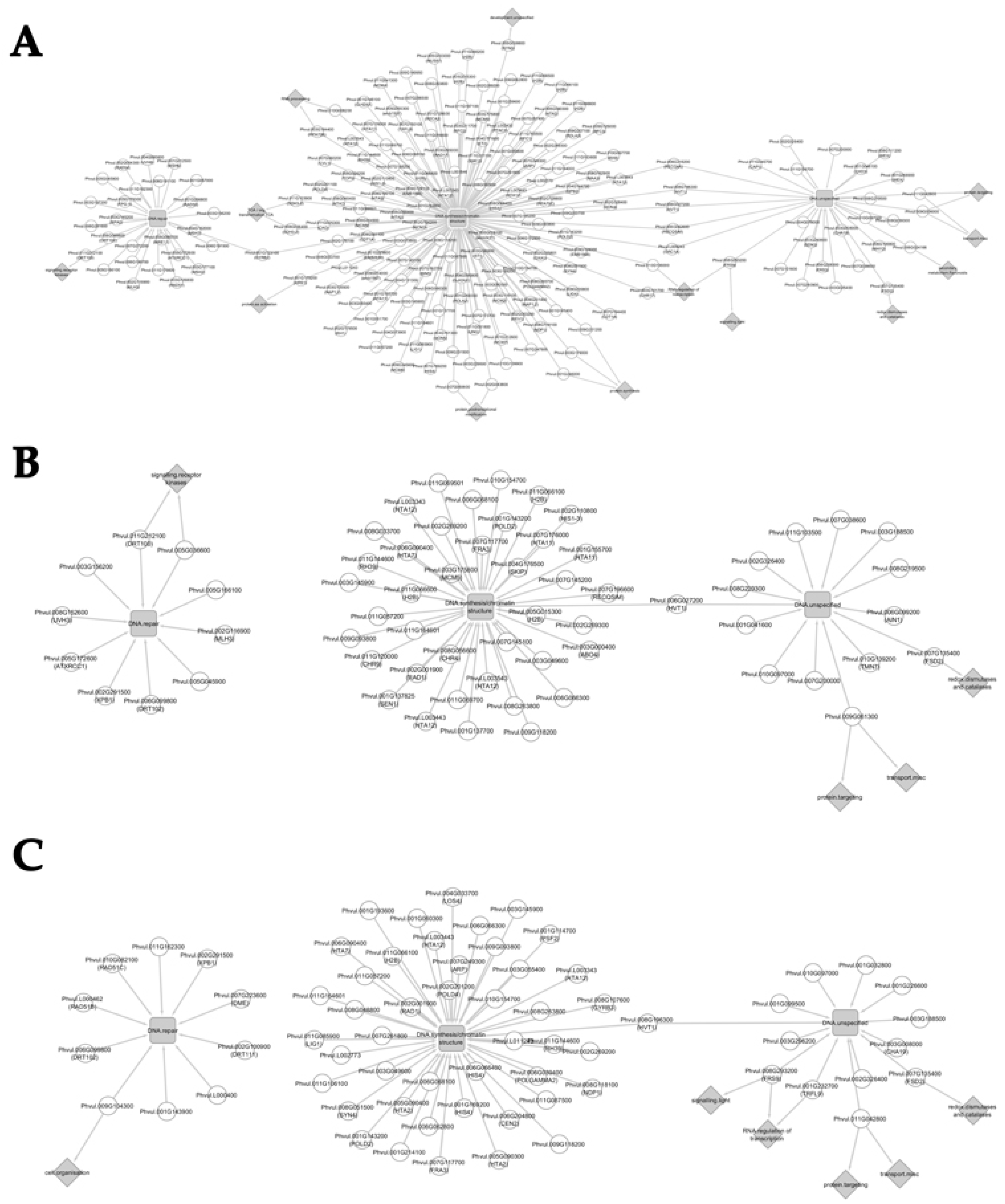
3.5. Network Analysis
3.5.1. Network Analysis from 10 to 20 Days after Anthesis
3.5.2. Network Analysis from 20 to 30 Days after Anthesis
3.5.3. Network Analysis from 30 to 40 Days after Anthesis
4. Discussion
4.1. Tight Control of DNA Damage Seems to Occur during Seed Development
4.2. Role of Chromatin and Chromatin Remodeling in DNA Damage Repair during Seed Development
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Waterworth, W.M.; Footitt, S.; Bray, C.M.; Finch-Savage, W.E.; West, C.E. DNA damage checkpoint kinase ATM regulates germination and maintains genome stability in seeds. Proc. Natl. Acad. Sci. USA 2016, 113, 9647–9652. [Google Scholar] [CrossRef] [PubMed]
- Roy, S. Maintenance of genome stability in plants: Repairing DNA double strand breaks and chromatin structure stability. Front. Plant Sci. 2014, 5, 487. [Google Scholar] [CrossRef] [PubMed]
- Manova, V.; Gruszka, D. DNA damage, and repair in plants—From models to crops. Front. Plant Sci. 2015, 6, 885. [Google Scholar] [CrossRef] [PubMed]
- Spampinato, C.P. Protecting DNA from errors and damage: An overview of DNA repair mechanisms in plants compared to mammals. Cell. Mol. Life Sci. 2017, 74, 1693–1709. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Abraham, R.T. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001, 15, 2177–2196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.; Cools, T.; De Veylder, L. Mechanisms psed by plants to cope with DNA damage. Annu. Rev. Plant Biol. 2016, 67, 439–462. [Google Scholar] [CrossRef] [PubMed]
- Yoshiyama, K.O. SOG1: A master regulator of the DNA damage response in plants. Genes Genet. Syst. 2015, 90, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Yoshiyama, K.O.; Kimura, S.; Maki, H.; Britt, A.B.; Umeda, M. The role of SOG1, a plant-specific transcriptional regulator, in the DNA damage response. Plant Signal. Behav. 2014, 9, e28889. [Google Scholar] [CrossRef] [PubMed]
- Tsukuda, T.; Fleming, A.B.; Nickoloff, J.A.; Osley, M.A. Chromatin remodelling at a DNA double-strand break site in Saccharomyces cerevisiae. Nature 2005, 438, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Parra, E.; Gutierrez, C. E2F regulates FASCIATA1, a chromatin assembly gene whose loss switches on the endocycle and activates gene expression by changing the epigenetic status. Plant Physiol. 2007, 144, 105–120. [Google Scholar] [CrossRef] [PubMed]
- Pagano, A.; Araújo, S.D.; Macovei, A.; Leonetti, P.; Balestrazzi, A. The seed repair response during germination: Disclosing correlations between DNA repair, antioxidant response, and chromatin remodeling in Medicago truncatula. Front. Plant Sci. 2017, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Donà, M.; Mittelsten Scheid, O. DNA damage repair in the context of plant chromatin. Plant Physiol. 2015, 168, 1206–1218. [Google Scholar] [CrossRef] [PubMed]
- Balestrazzi, A.; Confalonieri, M.; Macovei, A.; Donà, M.; Carbonera, D. Genotoxic stress and DNA repair in plants: Emerging functions and tools for improving crop productivity. Plant Cell Rep. 2011, 30, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Waterworth, W.M.; Bray, C.M.; West, C.E. The importance of safeguarding genome integrity in germination and seed longevity. J. Exp. Bot. 2015, 66, 3549–3558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelovici, R.; Galili, G.; Fernie, A.R.; Fait, A. Seed desiccation: A bridge between maturation and germination. Trends Plant Sci. 2010, 15, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Leprince, O.; Pellizzaro, A.; Berriri, S.; Buitink, J. Late seed maturation: Drying without dying. J. Exp. Bot. 2016, 68, erw363. [Google Scholar] [CrossRef] [PubMed]
- Murphy, T.M.; Belmonte, M.; Shu, S.; Britt, A.B.; Hatteroth, J. Requirement for abasic endonuclease gene homologues in Arabidopsis seed development. PLoS ONE 2009, 4, e4297. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Gao, Y.; Xu, H.; Zhang, Y.; Wang, J. A proteomic analysis of seed development in Brassica campestri L. PLoS ONE 2012, 7, e50290. [Google Scholar] [CrossRef] [PubMed]
- Leitão, S.T.; Dinis, M.; Veloso, M.M.; Šatović, Z.; Vaz Patto, M.C. Establishing the bases for introducing the unexplored portuguese common bean germplasm into the breeding world. Front. Plant Sci. 2017, 8, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Parreira, J.R.; Bouraada, J.; Fitzpatrick, M.A.; Silvestre, S.; Bernardes da Silva, A.; Marques da Silva, J.; Almeida, A.M.; Fevereiro, P.; Altelaar, A.F.M.; Araújo, S.S. Differential proteomics reveals the hallmarks of seed development in common bean (Phaseolus vulgaris L.). J. Proteom. 2016, 143, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Puryear, J.; Cairney, J. A simple and efficient method for isolating RNA from pine trees. Plant Mol. Biol. Rep. 1993, 11, 113–116. [Google Scholar] [CrossRef]
- Zawada, A.M.; Rogacev, K.S.; Müller, S.; Rotter, B.; Winter, P.; Fliser, D.; Heine, G.H. Massive analysis of cDNA Ends (MACE) and miRNA expression profiling identifies proatherogenic pathways in chronic kidney disease. Epigenetics 2014, 9, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Igor, A.Y.; Yakovlev, I.A.; Lee, Y.; Rotter, B.; Olsen, J.E.; Skrøppa, T.; Johnsen, Ø.; Fossdal, C.G. Temperature-dependent differential transcriptomes during formation of an epigenetic memory in Norway spruce embryogenesis. Tree Genet. Genomes 2014, 10, 355–366. [Google Scholar] [CrossRef]
- Eveland, A.L.; McCarty, D.R.; Koch, K.E. Transcript Profiling by 3′-untranslated region sequencing resolves expression of gene families. Plant Physiol. 2007, 146, 32–44. [Google Scholar] [CrossRef] [PubMed]
- Torres, T.T.; Metta, M.; Ottenwalder, B.; Schlotterer, C. Gene expression profiling by massively parallel sequencing. Genome Res. 2007, 18, 172–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef] [PubMed]
- De St. Groth, S.F.F. The evaluation of limiting dilution assays. J. Immunol. Methods 1982, 49, R11–R23. [Google Scholar] [CrossRef]
- Usadel, B.; Poree, F.; Nagel, A.; Lohse, M.; Czedik-Eysenberg, A.; Stitt, M. A guide to using MapMan to visualize and compare Omics data in plants: A case study in the crop species, Maize. Plant Cell Environ. 2009, 32, 1211–1229. [Google Scholar] [CrossRef] [PubMed]
- Thimm, O.; Bläsing, O.; Gibon, Y.; Nagel, A.; Meyer, S.; Krüger, P.; Selbig, J.; Müller, L.A.; Rhee, S.Y.; Stitt, M. MAPMAN: A user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 2004, 37, 914–939. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Nagel, A.; Herter, T.; May, P.; Schroda, M.; Zrenner, R.; Tohge, T.; Fernie, A.R.; Stitt, M.; Usadel, B. Mercator: A fast and simple web server for genome scale functional annotation of plant sequence data. Plant. Cell Environ. 2014, 37, 1250–1258. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2012, 40, D1178–D1186. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, I.; Choudhury, N.R.; Tuteja, N. Arabidopsis thaliana MCM3 single subunit of MCM2–7 complex functions as 3′ to 5′ DNA helicase. Protoplasma 2016, 253, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Masuda, H.P.; Ramos, G.B.A.; De Almeida-Engler, J.; Cabral, L.M.; Coqueiro, V.M.; Macrini, C.M.T.; Ferreira, P.C.G.; Hemerly, A.S. Genome based identification and analysis of the pre-replicative complex of Arabidopsis thaliana. FEBS Lett. 2004, 574, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, G.; van Gessel, N.; Köchl, F.; Hunn, L.; Schulze, K.; Maloukh, L.; Nogué, F.; Decker, E.L.; Hartung, F.; Reski, R. RecQ helicases function in development, DNA repair, and gene targeting in Physcomitrella patens. Plant Cell 2018, 30, 717–736. [Google Scholar] [CrossRef] [PubMed]
- Bagherieh-Najjar, M.B.; de Vries, O.M.H.; Kroon, J.T.M.; Wright, E.L.; Elborough, K.M.; Hille, J.; Dijkwel, P.P. Arabidopsis RecQsim, a plant-specific member of the RecQ helicase family, can suppress the MMS hypersensitivity of the yeast sgs1 mutant. Plant Mol. Biol. 2003, 52, 273–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans-Roberts, K.M.; Breuer, C.; Wall, M.K.; Sugimoto-Shirasu, K.; Maxwell, A. Arabidopsis thaliana GYRB3 does not encode a DNA gyrase subunit. PLoS ONE 2010, 5, e9899. [Google Scholar] [CrossRef] [PubMed]
- Bankaitis, V.A.; Mousley, C.J.; Schaaf, G. The Sec14 superfamily and mechanisms for crosstalk between lipid metabolism and lipid signaling. Trends Biochem. Sci. 2010, 35, 150–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, R. Arabidopsis FHY3/FAR1 gene family and distinct roles of its members in light control of Arabidopsis development. PLANT Physiol. 2004, 136, 4010–4022. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Yang, X.; Auman, D.; Makaroff, C.A. Expression of epitope-tagged SYN3 cohesin proteins can disrupt meiosis in Arabidopsis. J. Genet. Genomics 2014, 41, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Myouga, F.; Hosoda, C.; Umezawa, T.; Iizumi, H.; Kuromori, T.; Motohashi, R.; Shono, Y.; Nagata, N.; Ikeuchi, M.; Shinozaki, K. A heterocomplex of iron superoxide dismutases defends chloroplast nucleoids against oxidative stress and is essential for chloroplast development in Arabidopsis. Plant Cell Online 2008, 20, 3148–3162. [Google Scholar] [CrossRef] [PubMed]
- Hwang, M.G.; Kim, K.; Lee, W.-K.; Cho, M.H. AtTBP2 and AtTRP2 in Arabidopsis encode proteins that bind plant telomeric DNA and induce DNA bending in vitro. Mol. Genet. Genomics 2005, 273, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Ventura, L.; Donà, M.; Macovei, A.; Carbonera, D.; Buttafava, A.; Mondoni, A.; Rossi, G.; Balestrazzi, A. Understanding the molecular pathways associated with seed vigor. Plant Physiol. Biochem. 2012, 60, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.D.; Wu, Z.; Irizarry, R.A.; Leek, J.T. Sequencing technology does not eliminate biological variability. Nat. Biotechnol. 2011, 29, 572–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Li, N.; Guarnera, M.; Jiang, F. Quantification of plasma miRNAs by digital PCR for cancer diagnosis. Biomark. Insights 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.G.; Jeong, S.-Y.; Cho, K.-S. Comparison of droplet digital PCR and quantitative real-time PCR in mcrA-based methanogen community analysis. Biotechnol. Rep. 2014, 4, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Hindson, C.M.; Chevillet, J.R.; Briggs, H.A.; Gallichotte, E.N.; Ruf, I.K.; Hindson, B.J.; Vessella, R.L.; Tewari, M. Absolute quantification by droplet digital PCR versus analog real-time PCR. Nat. Methods 2013, 10, 1003–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukaide, M.; Sugiyama, M.; Korenaga, M.; Murata, K.; Kanto, T.; Masaki, N.; Mizokami, M. High-throughput and sensitive next-generation droplet digital PCR assay for the quantitation of the hepatitis C virus mutation at core amino acid 70. J. Virol. Methods 2014, 207, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Bharuthram, A.; Paximadis, M.; Picton, A.C.P.; Tiemessen, C.T. Comparison of a quantitative real-time PCR assay and droplet digital PCR for copy number analysis of the CCL4L genes. Infect. Genet. Evol. 2014, 25, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.; Duarte, S.; Tedesco, S.; Fevereiro, P.; Costa, R.L. Expression profiling of Castanea genes during resistant and susceptible interactions with the oomycete pathogen Phytophthora cinnamomi reveal possible mechanisms of immunity. Front. Plant Sci. 2017, 8, 515. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, K. Proteomics of medicago truncatula seed development establishes the time frame of diverse metabolic processes related to reserve accumulation. Plant Physiol. 2003, 133, 664–682. [Google Scholar] [CrossRef] [PubMed]
- Coelho, C.M.; Benedito, V. Seed development and reserve compound accumulation in common bean (Phaseolus vulgaris L.). Seed Sci. Biotechnol. 2008, 2, 42–52. [Google Scholar]
- Wang, W.-Q.; Liu, S.-J.; Song, S.-Q.; Møller, I.M. Proteomics of seed development, desiccation tolerance, germination and vigor. Plant Physiol. Biochem. 2015, 86, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Amiard, S.; Charbonnel, C.; Allain, E.; Depeiges, A.; White, C.I.; Gallego, M.E. Distinct roles of the ATR kinase and the Mre11-Rad50-Nbs1 complex in the maintenance of chromosomal stability in Arabidopsis. Plant Cell 2010, 22, 3020–3033. [Google Scholar] [CrossRef] [PubMed]
- Yoshiyama, K.O.; Kobayashi, J.; Ogita, N.; Ueda, M.; Kimura, S.; Maki, H.; Umeda, M. ATM-mediated phosphorylation of SOG1 is essential for the DNA damage response in Arabidopsis. EMBO Rep. 2013, 14, 817–822. [Google Scholar] [CrossRef] [PubMed]
- De Schutter, K.; Joubes, J.; Cools, T.; Verkest, A.; Corellou, F.; Babiychuk, E.; Van Der Schueren, E.; Beeckman, T.; Kushnir, S.; Inze, D.; et al. Arabidopsis WEE1 kinase controls cell cycle arrest in response to activation of the DNA integrity checkpoint. Plant Cell 2007, 19, 211–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amiard, S.; Depeiges, A.; Allain, E.; White, C.I.; Gallego, M.E. Arabidopsis ATM and ATR kinases prevent propagation of genome damage caused by telomere dysfunction. Plant Cell 2011, 23, 4254–4265. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Zhu, Y.; Dong, A.; Shen, W.-H. Histone H2A/H2B chaperones: From molecules to chromatin-based functions in plant growth and development. Plant J. 2015, 83, 78–95. [Google Scholar] [CrossRef] [PubMed]
- Bailly, C.; El-Maarouf-Bouteau, H.; Corbineau, F. From intracellular signaling networks to cell death: The dual role of reactive oxygen species in seed physiology. C. R. Biol. 2008, 331, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Naithani, S.; Nonogaki, H.; Jaiswal, P. Exploring crossroads between seed development and stress response. In Mechanism of Plant Hormone Signaling under Stress; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2017; Volume 2, pp. 415–454. [Google Scholar]
- Pagano, A.; Araújo, S.D.S.; Balestrazzi, A.; Dondi, D. Metabolic and gene expression hallmarks of seed germination uncovered by sodium butyrate in Medicago truncatula. Plant Cell Environ. 2018, 1–11. [Google Scholar] [CrossRef]
- Moldovan, G.-L.; Pfander, B.; Jentsch, S. PCNA, the maestro of the replication fork. Cell 2007, 129, 665–679. [Google Scholar] [CrossRef] [PubMed]
- Iyer, R.R.; Pluciennik, A.; Genschel, J.; Tsai, M.-S.; Beese, L.S.; Modrich, P. MutLα and proliferating cell nuclear antigen share binding sites on MutSβ. J. Biol. Chem. 2010, 285, 11730–11739. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Gu, L.; Li, G.-M. Distinct nucleotide binding/hydrolysis properties and molar ratio of MutSα and MutSβ determine their differential mismatch binding activities. J. Biol. Chem. 2009, 284, 11557–11562. [Google Scholar] [CrossRef] [PubMed]
- Maréchal, A.; Zou, L. RPA-coated single-stranded DNA as a platform for post-translational modifications in the DNA damage response. Cell Res. 2015, 25, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Aklilu, B.B.; Soderquist, R.S.; Culligan, K.M. Genetic analysis of the Replication Protein A large subunit family in Arabidopsis reveals unique and overlapping roles in DNA repair, meiosis and DNA replication. Nucleic Acids Res. 2014, 42, 3104–3118. [Google Scholar] [CrossRef] [PubMed]
- Kimura, S.; Tahira, Y.; Ishibashi, T.; Mori, Y.; Mori, T.; Hashimoto, J.; Sakaguchi, K. DNA repair in higher plants; photoreactivation is the major DNA repair pathway in non-proliferating cells while excision repair (nucleotide excision repair and base excision repair) is active in proliferating cells. Nucleic Acids Res. 2004, 32, 2760–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowan, B.A.; Oldenburg, D.J.; Bendich, A.J. RecA maintains the integrity of chloroplast DNA molecules in Arabidopsis. J. Exp. Bot. 2010, 61, 2575–2588. [Google Scholar] [CrossRef] [PubMed]
- Fujimori, N.; Suzuki, N.; Nakajima, Y.; Suzuki, S. Plant DNA-damage repair/toleration 100 protein repairs UV-B-induced DNA damage. DNA Repair (Amst) 2014, 21, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Paeng, S.K.; Nawkar, G.M.; Maibam, P.; Lee, E.S.; Kim, K.-S.; Lee, D.H.; Park, D.-J.; Kang, S.B.; Kim, M.R.; et al. The 1-Cys peroxiredoxin, a regulator of seed dormancy, functions as a molecular chaperone under oxidative stress conditions. Plant Sci. 2011, 181, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Boubriak, I.; McCready, S.; Osborne, D.J. DNA structure and seed desiccation tolerance. In Plant Desiccation Tolerance; Blackwell Publishing Ltd.: Oxford, UK, 2000; pp. 215–249. [Google Scholar]
- Giglia-Mari, G.; Zotter, A.; Vermeulen, W. DNA damage response. Cold Spring Harb. Perspect. Biol. 2011, 3, a000745. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.M.; Ye, X.; Hall, C.; Santos, H.; Ma, T.; Kao, G.D.; Yen, T.J.; Harper, J.W.; Adams, P.D. Coupling of DNA synthesis and histone synthesis in S phase independent of cyclin/cdk2 activity. Mol. Cell. Biol. 2002, 22, 7459–7472. [Google Scholar] [CrossRef] [PubMed]
- Feser, J.; Truong, D.; Das, C.; Carson, J.J.; Kieft, J.; Harkness, T.; Tyler, J.K. Elevated histone expression promotes life span extension. Mol. Cell 2010, 39, 724–735. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Liu, S.; Wang, J.; He, J.; Huang, H.; Zhang, Y.; Xu, L. ISWI proteins participate in the genome-wide nucleosome distribution in Arabidopsis. Plant J. 2014, 78, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Balestrazzi, A.; Donà, M.; Macovei, A.; Sabatini, M.E.; Pagano, A.; Carbonera, D. DNA repair and telomere maintenance during seed imbibition: Correlation of transcriptional patterns. Telomere Telomerase 2015, 2–5. [Google Scholar] [CrossRef]
- Ho, K.K.; Zhang, H.; Golden, B.L.; Ogas, J. PICKLE is a CHD subfamily II ATP-dependent chromatin remodeling factor. Biochim. Biophys. Acta-Gene Regul. Mech. 2013, 1829, 199–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, X.; Li, C.; Liang, Q.; Zhou, Y.; He, H.; Fan, L.-M. CHD3 chromatin-remodeling factor PICKLE regulates floral transition partially via modulating LEAFY expression at the chromatin level in Arabidopsis. Sci. China Life Sci. 2016, 59, 516–528. [Google Scholar] [CrossRef] [PubMed]
- Verdier, J.; Thompson, R.D. Transcriptional regulation of storage protein synthesis during dicotyledon seed filling. Plant Cell Physiol. 2008, 49, 1263–1271. [Google Scholar] [CrossRef] [PubMed]
- Zhai, L.; Xu, L.; Wang, Y.; Zhu, X.; Feng, H.; Li, C.; Luo, X.; Everlyne, M.M.; Liu, L. Transcriptional identification and characterization of differentially expressed genes associated with embryogenesis in radish (Raphanus sativus L.). Sci. Rep. 2016, 6, 21652. [Google Scholar] [CrossRef] [PubMed]
- Sreenivasulu, N.; Wobus, U. Seed-development programs: A systems biology-based comparison between dicots and monocots. Annu. Rev. Plant Biol. 2013, 64, 189–217. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yang, S.; Zhao, M.; Luo, M.; Yu, C.-W.; Chen, C.-Y.; Tai, R.; Wu, K. Transcriptional repression by histone deacetylases in plants. Mol. Plant 2014, 7, 764–772. [Google Scholar] [CrossRef] [PubMed]
- Mergner, J.; Schwechheimer, C. The NEDD8 modification pathway in plants. Front. Plant Sci. 2014, 5, 103. [Google Scholar] [CrossRef] [PubMed]
- Bostick, M. Related to ubiquitin 1 and 2 are redundant and essential and regulate vegetative growth, auxin signaling, and ethylene production in Arabidopsis. Plant Cell Online 2004, 16, 2418–2432. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Chen, Y.; Zhang, F.; Yang, C.-Y.; Wang, S.; Yu, X. RNF111-dependent neddylation activates DNA damage-induced ubiquitination. Mol. Cell 2013, 49, 897–907. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GENE ID | Gene Symbol | Description | Primer Forward Sequence (5’‒3’) | Primer Reverse Sequence (5’‒3’) | MGB Probe Sequence (5’‒3’) | Dye |
|---|---|---|---|---|---|---|
| Phvul.003G027100 | SOG1 | SUPPRESSOR OF GAMMA RESPONSE 1 | TGGGACAGTGAGTCACAGAA | GAGCATAAACAGAAAGACCAGGAT | CTGGGAGACTGGCTGTGGAGGAGAT | FAMTM |
| Phvul.006G003000 | ATM | ATAXIA-TELANGIECTASIA MUTATED | TGGACTCAGATCAGGCATTGA | CACCAAAATCAGTGTCACCTCTT | AGCAGGCGGCAATGGATGTGGTT | FAMTM |
| Phvul.006G095800 | G-H2AX | GAMMA HISTONE VARIANT H2AX | GGTGAGGAATGATGAGGAACTG | ACTCTTGTGAAGCAGATCCAA | CCGTTCGCAATGGTGACAGACCCC | FAMTM |
| Phvul.005G085700 | MRE11 | DNA REPAIR AND MEIOSIS PROTEIN (MRE11) | CCACCTCGGGTATATGGAGAA | AAATCTCTTCAAAGGCGTGGAA | ATGAGGTGCGCCGCCACGACT | FAMTM |
| Phvul.001G266800 | RAD50 | DNA REPAIR-RECOMBINATION PROTEIN (RAD50) | TGATGGTATGCGGCAAATGTTT | GCTGAACTAGTAGCCTTCACTCTT | TGCCCTTGCTGTGAACGCCCT | VICTM |
| Phvul.008G242800 | NBS1 | NIJMEGEN BREAKAGE SYNDROME 1 | CAAGGTTGATGATAATGAAACTGGAA | GAGTGTGTGCCTTTCTGAAACAT | TGCTGTCTGGTGCAGTGCTTACGCT | VICTM |
| Phvul.001G204900 | WEE1 | WEE1 KINASE HOMOLOG | CTCATTCCTCTCAACCAACCA | GTGAGCACAACGCACGAT | CCTCCGTTTCCTGCTTCCAGAACCC | VICTM |
| Phvul.003G135500 | NAP1;2 | NUCLEOSOME ASSEMBLY PROTEIN 1;2 | CTTTCACCTCTGCAATGAGTAAC | CCGCTCTATTTTCCTCGTTGA | AGGACACCTTCAACGTCGCCGATCT | VICTM |
| Pooled Sample ID | Raw Reads | Cleaned Reads | Mapped Reads | % Mapped Reads |
|---|---|---|---|---|
| 10 DAA | 10880000 | 8000000 | 7818197 | 97.73% |
| 20 DAA | 10767653 | 7876850 | 7633075 | 96.91% |
| 30 DAA | 11648490 | 8477795 | 8296129 | 97.86% |
| 40 DAA | 8255161 | 5772840 | 5518643 | 95.60% |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parreira, J.R.; Balestrazzi, A.; Fevereiro, P.; Araújo, S.D.S. Maintaining Genome Integrity during Seed Development in Phaseolus vulgaris L.: Evidence from a Transcriptomic Profiling Study. Genes 2018, 9, 463. https://doi.org/10.3390/genes9100463
Parreira JR, Balestrazzi A, Fevereiro P, Araújo SDS. Maintaining Genome Integrity during Seed Development in Phaseolus vulgaris L.: Evidence from a Transcriptomic Profiling Study. Genes. 2018; 9(10):463. https://doi.org/10.3390/genes9100463
Chicago/Turabian StyleParreira, José Ricardo, Alma Balestrazzi, Pedro Fevereiro, and Susana De Sousa Araújo. 2018. "Maintaining Genome Integrity during Seed Development in Phaseolus vulgaris L.: Evidence from a Transcriptomic Profiling Study" Genes 9, no. 10: 463. https://doi.org/10.3390/genes9100463