Profiling DNA Methylation Based on Next-Generation Sequencing Approaches: New Insights and Clinical Applications




1
Cancer Biology and Epigenetics Group, IPO Porto Research Center (CI-IPOP), Portuguese Oncology Institute of Porto (IPO Porto), Rua António Bernardino Almeida, 4200-072 Porto, Portugal
2
Genetics, Department of Pathology, Faculty of Medicine, University of Porto, 4200-319 Porto, Portugal
3
I3S—Instituto de Investigação e Inovação em Saúde, Universidade do Porto, 4200-135 Porto, Portugal
4
Department of Pathology, Portuguese Oncology Institute of Porto (IPO Porto), 4200-072 Porto, Portugal
5
Department of Pathology and Molecular Immunology, Institute of Biomedical Sciences Abel Salazar (ICBAS)—University of Porto, 4050-313 Porto, Portugal
*
Author to whom correspondence should be addressed.
Genes 2018, 9(9), 429; https://doi.org/10.3390/genes9090429
Submission received: 23 May 2018
/
Revised: 28 July 2018
/
Accepted: 20 August 2018
/
Published: 23 August 2018
(This article belongs to the Special Issue Emerging Applications for Next Generation Sequencing)
Abstract
:DNA methylation is an epigenetic modification that plays a pivotal role in regulating gene expression and, consequently, influences a wide variety of biological processes and diseases. The advances in next-generation sequencing technologies allow for genome-wide profiling of methyl marks both at a single-nucleotide and at a single-cell resolution. These profiling approaches vary in many aspects, such as DNA input, resolution, coverage, and bioinformatics analysis. Thus, the selection of the most feasible method according with the project’s purpose requires in-depth knowledge of those techniques. Currently, high-throughput sequencing techniques are intensively used in epigenomics profiling, which ultimately aims to find novel biomarkers for detection, diagnosis prognosis, and prediction of response to therapy, as well as to discover new targets for personalized treatments. Here, we present, in brief, a portrayal of next-generation sequencing methodologies’ evolution for profiling DNA methylation, highlighting its potential for translational medicine and presenting significant findings in several diseases.
1. Introduction
DNA methylation patterns’ changes have been widely studied and constitute the most well understood epigenetic modification. The addition of a methyl group at the 5′ position of the cytosine residue proved to be an essential mechanism in both gene expression and chromatin structure regulation [1]. The functional presence of 5mC (5-methylcytosine) in gene promoters is generally associated with transcriptional repression due to structural chromatin alterations, while the absence is linked with transcriptional activity. Gene body methylation also plays a major role in repetitive DNA elements’ silencing and alternative splicing [2,3]. DNA methylation has been associated with several biological processes such as genomic imprinting, transposon inactivation, stem cell differentiation, transcription repression, and inflammation [4]. DNA methylation profiles can also be inherited through cell division and sometimes through generations [5]. Since methyl marks play a very relevant role in both physiologic and pathologic conditions, the significance of profiling DNA methylation to answer biological questions has been highlighted. Moreover, uncovering of DNA methylation genomic regions is appealing to translational research because methyl sites are modifiable by pharmacologic intervention [6] and are easy to measure in liquid biopsies through cost-effective methods [7]. However, efforts to examine epigenetic modifications in health and disease have been hindered by the lack of high-throughput and quantitatively accurate approaches, until recently.
Over the last years, numerous methods were developed to map 5mC providing a genome-wide coverage of DNA methylation changes. This improvement in DNA methylation analysis techniques was only possible due to the advances of various profiling approaches, both experimental and computational [8,9,10]. Epigenetics was one of the first molecular fields capitalizing next-generation sequencing (NGS) methodologies to its favor, providing a comprehensive and unbiased view of the epigenome and also releasing researchers from content-limited microarray platforms [11]. Next-generation sequencing technology has brought unprecedented advancement to epigenomic research, particularly in DNA methylation landscape. Compared to array-based technologies, NGS made possible deep sequencing in a short time (from one to few days), producing billions of short DNA samples known as reads (ranging from 50–400 nucleotides) and providing better coverage of all possible methylation sites in the human genome [12]. The arrival of NGS technologies allows for each of the three billion bases in the human genome to be sequenced multiple times, enabling high depth reading to deliver accurate data and insight into unexpected DNA variations [13]. Thanks to massively parallel sequencing it is now possible to recognize methylation status on a large scale and at a single-base resolution [9].
A complete characterization of the methylome and the dynamic changes that occur within may serve as an accurate diagnostic, prognostic, and predictive tools. This mini-review aims to provide a brief overview of NGS tools that might be used in translational medical research, discussing the main advantages and limitations of those technologies and making considerations about the advantages and limitations of each method.
2. DNA Methylation Profiling
Pyrosequencing, methylation-specific polymerase chain reaction (PCR), and direct Sanger sequencing have been the most widely used methods for analysis of targeted regions, such as a promoter region of a single gene or a CpG (Cytosine-phosphate-Guanine) island. Although highly useful, the limitations of these techniques include low quantitative accuracy, short read length, and low sample throughput.
A plethora of new methods are currently used to investigate 5mC epigenetic landscape in DNA. Microarray hybridization was one of the first technologies escalating the DNA methylation studies to a genome-wide level. Notably, the methylation arrays are cost-effective tools which do not require large amounts of input DNA, enabling simultaneous analysis of several samples. However, the coverage is highly dependent on the array design [14]. Next-generation sequencing platforms are now emerging, allowing for massive analysis of the methylation status of almost every CpG site and construction of DNA methylation’s genomic maps at a single base resolution [15]. Nevertheless, most of the approaches to 5mC analysis still have the restrictions of being density-biased, deficient in robustness and consistency, or incapable of analyzing 5mC specifically [16]. Hence, for DNA methylation studies involving clinical research, the combination of next-generation sequencing and methylation arrays may constitute a powerful approach as discovery and screening tools, respectively [9].
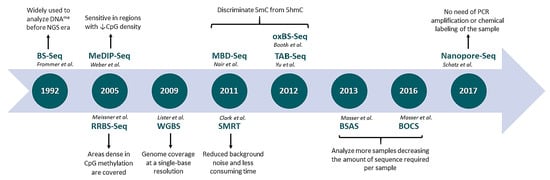
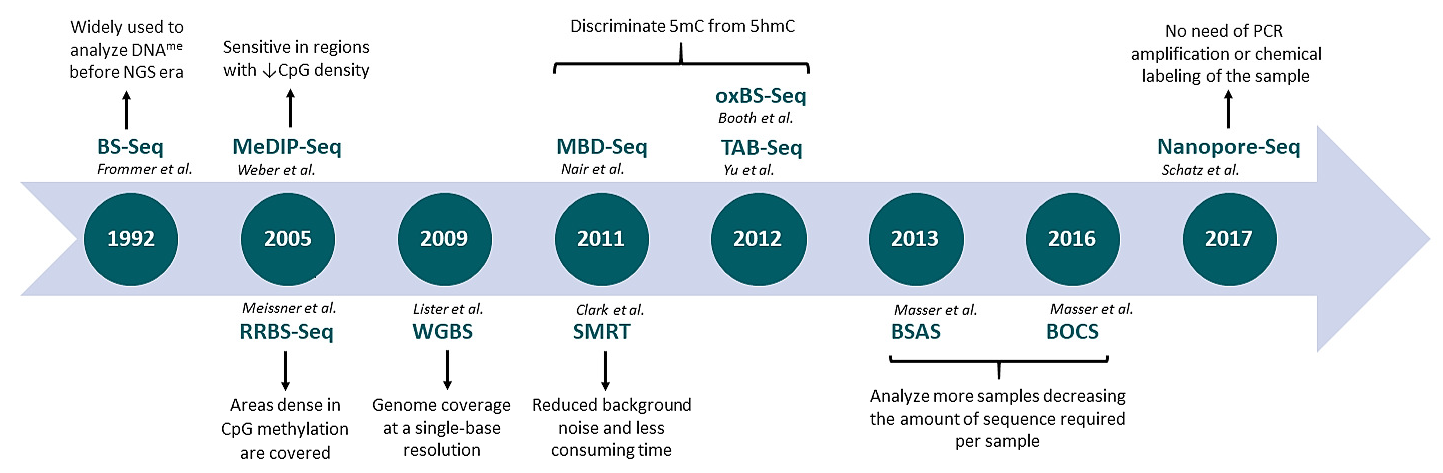
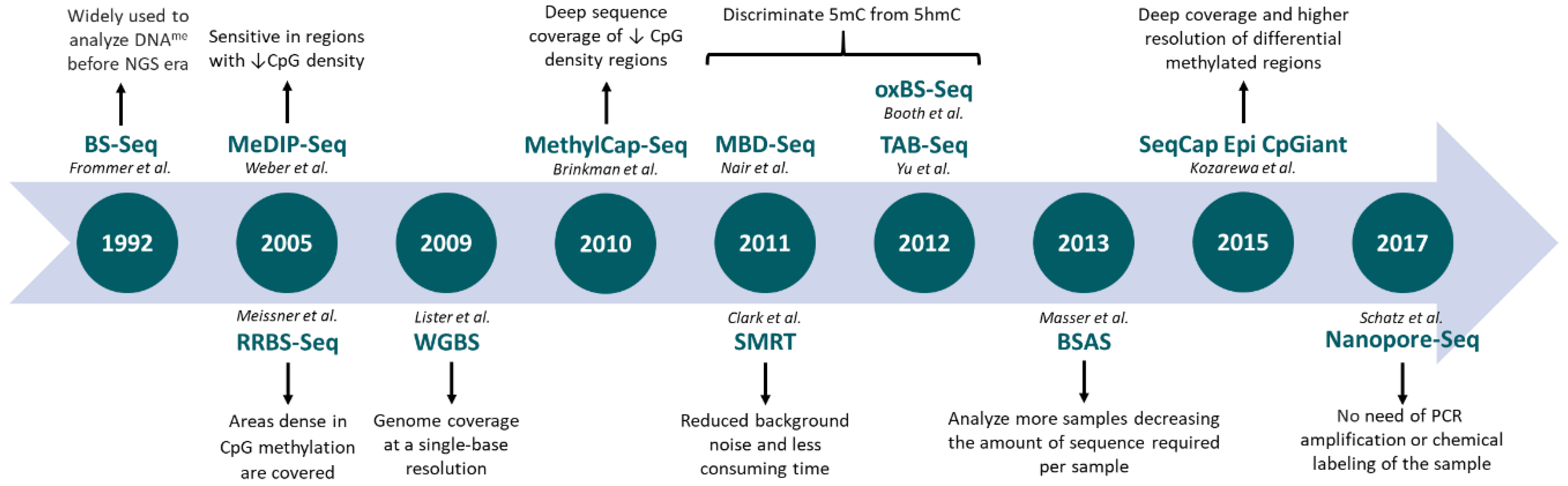
A detailed characterization of the most commonly used genome-wide techniques is depicted in Table 1. An historic overview of NGS-based methods applied to epigenomics is shown in Figure 1.
2.1. Affinity Enrichment-Based Methods
This strategy uses antibodies (methylated DNA immunoprecipitation or MeDIP-Seq) and methylated-CpG binding proteins (MBD-Seq) to pull down the genomic regions that are methylated for sequencing. The unmethylated genomic fraction is washed away and the methylation-enriched portion is then collected and sequenced [17,18]. Although enrichment techniques are biased towards hypermethylated areas, which are preferentially or more effectively captures, they are particularly useful for characterizing enriched CpG regions (CpG islands and promoter regions).
The main limitations of these approaches are the limited quantification across regions and lack of base-specific data analysis, which ultimately diminishes the insight that could be gathered from them. Thus, these affinity-based methodologies require substantial experimental and bioinformatics adjustments [9]. Sequence data are more commonly analyzed using count-based models, but this statistical method should be avoided in large group samples’ analysis since the biological variability is ignored and more false positives are generated. In this context, a beta-binomial model works more favorably, because it is flexible enough to capture both the technical (coverage) and biological (methylation level) variability [19,20].
2.2. Restriction Enzymes-Based Methods
Restriction-enzyme-based approaches take advantage of restriction enzymes (MspI) that are able to cleave the recognition sequence at the site of DNA methylation (CCGG motifs), thus, 5mC can be identified in selected sequences [21]. This is the most time and cost-effective sequencing method and require minute amounts of DNA [22]. However, only lower weight fragments, between 40–220 bp, are suitable for sequencing, despite a wide range of lengths resulting from digestion. The primary limitation to this method is the inability to tune coverage regions of interest due to the dependence on the restriction sites location in the genome, leading to a lack of applicability in genes with sparse CCGG motifs [23].
2.3. Bisulfite Conversion-Based Methods
In bisulfite sequencing (BS-Seq), denatured DNA is subjected to bisulfite treatment during which the unmodified cytosine is converted to uracil, but a methylated cytosine remains unchanged, thus allowing base resolution detection of cytosine methylation [24,25]. The bisulfite treatment makes methylation sequencing data processing challenging because of C → T conversion. Thus, it is important to bear in mind that bisulfite sequence reads are not complementary to the reference genome and, therefore, special alignment tools are usually required [19]. In this particular case another level of complexity is added because every given thymine could either be a genuine genomic thymine or a converted unmethylated cytosine, rendering conventional alignment tools, such as Bowtie or BWA (Burrows-wheeler aligner), unsuitable [26]. However, several new computational tools have been developed to address this issue, namely BSMAP [27] and Segemehl [28], which enumerate all C to T combinations in the read, or BISMARK [29] and BS-Seeker [30], which convert all C to T in both sequenced reads and genome reference prior to alignment.
Whole genome bisulfite sequencing (WGBS) is the most informative but also the most expensive base resolution technology since whole genome is targeted by this method. For WGBS, genomic DNA libraries are created and subsequently bisulfite converted, sequenced, and mapped back to the reference genome. Although BS-seq is the most direct assay and displays the highest resolution for methylation detection, this methodology is only used for seeking specific questions in which comprehensive DNA methylation profile is required [31].
Over the last years, numerous WGBS studies demonstrated that the majority of CpG sites are equally methylated and only a small portion of these regions depicts variable DNA methylation, the so-called differentially methylated regions (DMR). Thus, development of new sequencing approaches, more useful for studies testing specific regions of interest and capable of focus only in DMR are needed to efficiently validate putative candidate loci across a large number of samples. In this context, Masser et al. developed an approach, termed bisulfite amplicon sequencing (BSAS), for hypothesis driven and focused absolute DNA methylation analysis [32]. With the onset of NGS platforms it was possible to sequence multiple samples in parallel in one run using multiplexed amplicon-based NGS [33]. This new methodology is highly sensitive and integrates DNA barcoding into the library construction process, so that many samples (from 96 up to 384 different samples) can be pooled to fully use NGS capacity [34].
2.4. Oxidative Bisulfite Conversion-Based Methods
One of the most recent developments in epigenomic profiling was the discovery of alternative cytosine modifications with relevant functional biologic roles, including 5-hydroxymethylcytosine (5hmC), 5-carboxylcytosine (5caC), and 5-formylcytosine (5fC), Ito et al. [35], which are postulated to be involved in the process of DNA demethylation [36]. Conventional bisulfite sequencing is not able to distinguish between 5mC and 5hmC and the methylation profile resulting from this methodology includes the sum of both epi-marks [37]. Indeed, when specific techniques were applied for hydroxymethylation discrimination, a quarter of what was previously assumed as methylation was, in fact, hydroxymethylation [38]. Thus, the development of new methods for DNA profiling based on NGS that discriminate between methylation and hydroxymethylation was imperative. A modified bisulfite sequencing approach—oxidative bisulfite sequencing (Ox-BS)—includes a selective oxidative step that deprotects hydroxymethylation and converts 5hmC to 5fC, which, after bisulfite treatment, becomes a uracil [39,40]. Then a subtractive approach between bisulfite sequencing (BS) and Ox-BS libraries allows for methylation and hydroxymethylation quantification at single-base resolution. The main drawbacks of Ox-BS are the oxidative degradation of DNA and longer bisulfite treatment required for complete 5fC deamination [41].
An alternative method for 5hmC detection is Tet-assisted bisulfite sequencing (TAB-Seq), which might be used for whole genome or locus-specific sequencing [42]. This technique encompasses β-glucosyltransderase-mediated protection of 5hmC followed by Tet1-mediated oxidation of 5mC to 5caC. Then, bisulfite treatment and subsequent PCR amplification take place and 5caC, as well as cytosine, are converted to thymine, whereas 5hmC reads as cytosine [43]. Compared with OxBS-Seq, TAB-Seq directly reads 5hmC and the treatment methodology incurs less DNA damage. Nevertheless, TAB-Seq entails highly active Tet protein to enable efficient conversion of 5mC into 5caC [44].
2.5. Capture-Based Methods
The ability to capture and sequence large contiguous DNA fragments represents a significant advance towards comprehensive characterization of complex genomic regions. Capture-based DNA sequencing is advantageous not only because it is more cost-effective, as it facilitates higher sample throughput than whole genome sequencing, but also because it improves accuracy by optimizing the read depth coverage and by reducing the complexity of the DNA to be sequenced [45].
Several studies have highlighted the importance of differential methylation outside CpG islands as disease-associated determinants [46,47,48]. Thus, a new methylation-capture method—MethylCap-seq—was developed aiming at deep sequence coverage of lower CpG density regions. This technique is based on DNA methylation capture with MBD (methyl-CpG binding) domain of MeCP2 (methyl-CpG binding protein 2), which is advantageous for a highly controlled and stepwise elution and stratification of methylated DNA fragments according to methyl-CpG density. Currently, MethylCap-seq has been efficiently robotized enabling high reproducibility among numerous samples [18].
Another common bisulfite methylation sequencing method is SeqCap Epi CpGiant which is based on capture of bisulfite-converted DNA. This is also a target-enrichment protocol for genomic regions where methylation is known to impact gene regulation. It allows for sequencing of pre-selected regions with a genome coverage of approximately 80 million bases and 5 million CpG sites [19].
2.6. Third-Generation Sequencing
This recent technology allows for DNA modifications analysis without previous chemical conversion. Although conversion-based sequencing is currently the most used NGS methodology, this has some flaws, including difficulties in conversion efficiency control and in accurate alignment of altered sequences to their reference genome. Clark et al. [49] established an alternative approach, consisting on single-molecule real-time (SMRT) DNA sequencing to recognize modified DNA bases in the DNA template directly. This new methodology is based on changes in the kinetics of DNA polymerase (stretches of fluorescent signals represent the dynamics of DNA polymerization) during the occurrence of the modified bases [49,50]. In the same line, another method based on nanopore sequencers allows for direct read of different modifications on DNA bases. Nanopore sequencing uses pores though which nucleic acid strands are pulled, and the consequent ionic pattern reveals the nucleotide sequence, including modifications [51,52]. Although these technologies are still in the development phase, they seem promising for future methylome profiling analysis.
3. New Insights from Next-Generation Sequencing on Methylome Analysis: Strengths and Weaknesses
Next-generation sequencing revolutionized the methylome analysis contributing with a variety of new methods which expanded knowledge and characterization of differentially methylated DNA regions (Table 2). Especially in clinical research, DNA methylation status assessment through NGS in clinical samples might provide relevant information on diagnosis and prognosis. Furthermore, pharmacoepigenomics is another promising clinical epigenetic field in which the use of NGS might be of great importance, since the methylation status at specific candidate gene promoters in certain tumors predicts the likelihood of clinical response to treatment [53,54].
Considering the methylome coverage requirements, sample throughput, and resources available, the current range of technologies is able to meet most needs. Over the last few years, a considerable increase in the stability, throughput, and quality of NGS has been attained. These massive parallel sequencing technologies allow for comprehensive interrogation of genomes without prior knowledge of sequence or annotation. The relative low amounts of starting material reduced the errors and bias caused by sample preparation and amplification [63]. Next-generation reads are generated from fragmented and adapter-ligated DNA libraries that have never been subjected to conventional vector-based cloning, enabling to circumvent some of the sequencing bias of cloned DNA sequences that affect genome identification in sequencing projects [16]. Furthermore, high sensitivity, specificity, and scalability make this technology a powerful tool for the search of novel epigenetic biomarkers [9]. Additionally, signal quantification from sequence-based approaches focus on counting sequence tags rather than relative measures between samples, enabling an unlimited, fully-quantitative result [11]. Lastly, the increase in the amount of data generated per run and the decrease in reagent costs resulted in a higher cost-effectiveness of NGS for methylome profiling [63].
Currently, NGS is used to characterize several types of cancer and enabled the construction of large-scale databases such as The Cancer Genome Atlas (TCGA) (http://cancergenome.nih.gov/) and International Cancer Genome Consortium (http://icgc.org). These databases contain hundreds of cancer profiles based on whole-genome sequencing, gene expression and protein profiling, RNA sequencing, methylome analysis and copy number variation [50].
However, to best suit clinical purposes, targeted NGS panels are now emerging, assessing specific gene mutations that may assist in diagnosis and selection of targeted therapy. With targeted sequencing, a subset of genomic regions are isolated and sequenced, allowing researchers to focus time, costs, and data analysis on specific areas of interest and enabling sequencing at much higher coverage levels (500–1000× higher). For assay development, amplicon-based methods have been preferentially used because of their simplified workflow and small amounts of input DNA required. However, capture sequencing has emerged as an alternative approach because of high testing accuracy with respect to sequencing complexity and uniformity of coverage [64].
The main drawback for NGS in the clinical setting is the infrastructure required, including computer capacity and storage, and, importantly, expert bioinformatician support to comprehensively analyze and interpret the vast amount of data that is generated [13]. Moreover, the effective interpretation of datasets generated in different laboratories with common DNA profiling techniques requires the adoption of standards in both experimental and computational methods to allow for meaningful comparisons between experiments [10]. Importantly, quality control matrices and procedures should be adopted during base resolution methylome profiling. Methylation sequencing quality control includes reads alignment, methylation scoring, methylation heterogeneity assessment, genomic features annotation, data visualization, and determination of differentially methylated cytosines. These quality control steps are integrated into analytic pipelines, such as MethylQA or BSeQC tools [65,66].
4. Non-Invasive DNA Methylation Detection Using Next-Generation Sequencing: Technical Advances and Challenges
Until a few years ago, the majority of DNA methylation studies investigated genome methylation status using tissue-extracted DNA. However, because the tissue availability depends on invasive procedures, it limits it usefulness and, especially in biopsies, heterogeneity might be underestimated due to sampling limitations. Heterogeneous methylation found in tissues makes the fraction of methylation segments difficult to quantify with precision, compromising its use as a biomarker [67].
Currently, sequencing methylated DNA in a single-cell is now possible using a variety of experimental approaches, providing provides further insights into the links between cell’s phenotype and genotype. Parallel measurement of single-cell epigenomes might improve our understanding of normal developmental and disease processes [68]. As previously mentioned, DNA methylation commonly correlates with gene expression variations in mammals. Thus, to explore the cause-consequence basis for this association, measurements of both DNA methylation and transcript expression for the same single-cells is mandatory to uncover the dynamics of heterogeneous cell populations [69]. Currently several methods, including methylome and transcriptome sequencing from single-cell (scM&T-seq) and single-cell triple omics sequencing (scTrio-seq), allow for the consideration of whether DNA methylation extent of different functional elements in the genome impacts on the expression levels of genes in single cells [70,71,72].
Researchers are now investing in the development of less invasive and more accessible approaches to complement, and eventually substitute, tissue DNA analysis. Bodily fluids, such as plasma, serum, urine, or even saliva, often harbor increased cell-free DNA levels, which may be potentially detected using massive parallel sequencing [73]. Thus, NGS constitutes a promising tool for clinical biomarkers’ research due to its high sensitive and time/cost efficiency [74]. The use of NGS in liquid biopsies showed great potential for molecular testing and currently this methodology is largely applied for clinically relevance hotspot mutations detection. However, these types of genetic mutations are only detected in a small subset of patients, limiting its use for early diagnosis strategies [75,76,77]. With the emergence of epigenetics, aberrant DNA methylation profiles were shown to be early and common events during illness development, enabling a more robust detection and higher sensitivity in diagnosis [1,78]. Thus, escalating liquid biopsies molecular testing using NGS technologies to identify epigenetic signatures will potentially increase the diagnostic yield and clinical usefulness [79]. Furthermore, different tissue types may share similar genotypes but display different methylation profiles, and, thus, type-specific methylation signatures can potentially be used to identify the tissue of origin [80,81].
Regarding applications to clinical practice, NGS has been used to determine genome-wide profiles in serum or plasma from different cancers [82,83,84]. The single base resolution is attractive because it allows for precise mapping of relevant disease-specific sites. However, the combination of high cost of sequencing entire genomes and the large number of samples needed to provide adequate statistical power makes WGBS not economically viable as a screening tool for disease association studies at this time. Currently, a commonly used cost-effective alternative is methylation-capture followed by NGS [85]. Nevertheless, the application of these protocols to the methylation fraction of cell-free DNA still requires overcoming several technical challenges. Firstly, cell-free DNA occurs at a very low concentration levels leading to non-specific bindings during sample capture process and constitutes a critical limitation for this methodology. A second drawback results from the methylation enrichment step, which recovers a minimal fraction of the total DNA input, reducing the amount of DNA available for NGS library preparation [86,87]. Furthermore, ameliorate the quality of cell-free DNA extraction procedures, improving target candidate’s selection to increase sequencing coverage and find suitable reference DNA methylomes might speed-up the use of NGS technologies in non-invasive epigenetic tests to assist in diagnosis and prognostication.
5. Conclusions
The epigenetic community rapidly combined NGS with established DNA methylation capture methods enabling the improvement of the methylome analysis. Hence, in the last years, NGS became an effective tool for DNA methylation profiling at a single-base level and at relatively affordable price. Furthermore, the use of NGS contributed to increased knowledge on differentially methylated DNA regions and the discovery of new gene regulatory elements involved in the epigenetic machinery. These massive-parallel technologies offer great promise for decoding the nature and patterns of DNA modifications, as well as their implications in the various pathologic and physiologic processes. There are, however, other applications of high-throughput sequencing technologies in base modification. Information about DNA methylation patterns and distribution in the human genome is also important for personalized epigenomic-based therapy development.
Author Contributions
Conception and design: D.B.-S. and C.J.M.; Revision and editing: C.J.M., R.H. and C.J.
Funding
This research was funded by Research Center—Portuguese Oncology Institute of Porto (CI-IPO-FB-GEBC-2018 and FCT (POCI-01-0145-FEDER-29043). D.B.-S. is a research fellow from CI-IPOP (BI-GEBC2018/UID/DTP/00776/POCI-01-0145-FEDER-006868), and C.J.M. is a FCT Investigador (FCT contract IF/00047/2012).
Conflicts of Interest
The authors declare they have no competing interest.
References
- Esteller, M. Epigenetics in cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef] [PubMed]
- Laurent, L.; Wong, E.; Li, G.; Huynh, T.; Tsirigos, A.; Ong, C.T.; Low, H.M.; Sung, K.W.K.; Rigoutsos, I.; Loring, J. Dynamic changes in the human methylome during differentiation. Genome Res. 2010, 20, 320–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maunakea, A.K.; Chepelev, I.; Cui, K.; Zhao, K. Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 2013, 23, 1256. [Google Scholar] [CrossRef] [PubMed]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Klosin, A.; Lehner, B. Mechanisms, timescales and principles of trans-generational epigenetic inheritance in animals. Curr. Opin. Genet. Dev. 2016, 36, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Issa, J.-P.J.; Kantarjian, H.M. Targeting DNA methylation. Clin. Cancer Res. 2009, 15, 3938–3946. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, E.; Ulz, P.; Geigl, J.B. Circulating tumor DNA as a liquid biopsy for cancer. Clin. Chem. 2015, 61, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Heyn, H.; Esteller, M. DNA methylation profiling in the clinic: Applications and challenges. Nat. Rev. Genet. 2012, 13, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Soto, J.; Rodriguez-Antolin, C.; Vallespín, E.; De Castro Carpeño, J.; De Caceres, I.I. The impact of next-generation sequencing on the DNA methylation–based translational cancer research. Transl. Res. 2016, 169, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Hirst, M.; Marra, M.A. Next generation sequencing based approaches to epigenomics. Brief. Funct. Genom. 2010, 9, 455–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurd, P.J.; Nelson, C.J. Advantages of next-generation sequencing versus the microarray in epigenetic research. Brief. Funct. Genom. Proteom. 2009, 8, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Ecker, J.R. Finding the fifth base: Genome-wide sequencing of cytosine methylation. Genome Res. 2009, 19, 959–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behjati, S.; Tarpey, P.S. What is next generation sequencing? Arch. Dis. Child.-Educ. Pract. 2013, 98, 236–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yong, W.-S.; F.-Hsu, M.; Chen, P.-Y. Profiling genome-wide DNA methylation. Epigenet. Chromatin 2016, 9, 26. [Google Scholar] [CrossRef] [PubMed]
- Laird, P.W. Principles and challenges of genome-wide DNA methylation analysis. Nat. Rev. Genet. 2010, 11, 191. [Google Scholar] [CrossRef] [PubMed]
- Anandhakumar, C.; Kizaki, S.; Bando, T.; Pandian, G.N.; Sugiyama, H. Advancing small-molecule-based chemical biology with next-generation sequencing technologies. Chembiochem 2015, 16, 20–38. [Google Scholar] [CrossRef] [PubMed]
- Serre, D.; Lee, B.H.; Ting, A.H. MBD-isolated Genome Sequencing provides a high-throughput and comprehensive survey of DNA methylation in the human genome. Nucleic Acids Res. 2009, 38, 391–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinkman, A.B.; Simmer, F.; Ma, K.; Kaan, A.; Zhu, J.; Stunnenberg, H.G. Whole-genome DNA methylation profiling using MethylCap-seq. Methods 2010, 52, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Cunningham, J.; Slager, S.; Kocher, J.P. Base resolution methylome profiling: Considerations in platform selection, data preprocessing and analysis. Epigenomics 2015, 7, 813–828. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Baheti, S.; Sun, Z. Statistical method evaluation for differentially methylated CpGs in base resolution next-generation DNA sequencing data. Brief. Bioinf. 2016, 19, 374–386. [Google Scholar] [CrossRef] [PubMed]
- Maunakea, A.K.; Nagarajan, R.P.; Bilenky, M.; Ballinger, T.J.; D’souza, C.; Fouse, S.D.; Johnson, B.E.; Hong, C.; Nielsen, C.; Zhao, Y.; et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 2010, 466, 253. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Sun, J.; Wu, H.; Liu, S.; Wang, J.; Wu, B.; Huang, S.; Li, N.; Wang, J.; Zhang, X. Systematic assessment of reduced representation bisulfite sequencing to human blood samples: A promising method for large-sample-scale epigenomic studies. J. Biotechnol. 2012, 157, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Smith, Z.D.; Bock, C.; Boyle, P.; Gnirke, A.; Meissner, A. Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nat. Protoc. 2011, 6, 468. [Google Scholar] [CrossRef] [PubMed]
- Hayatsu, H. Discovery of bisulfite-mediated cytosine conversion to uracil, the key reaction for DNA methylation analysis—A personal account. Proc. Jpn. Acad. Ser. B 2008, 84, 321–330. [Google Scholar] [CrossRef]
- Frommer, M.; McDonald, L.E.; Millar, D.S.; Collis, C.M.; Watt, F.; Grigg, G.W.; Molloy, P.L.; Paul, C.L. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. USA 1992, 89, 1827–1831. [Google Scholar] [CrossRef] [PubMed]
- Adusumalli, S.; Mohd Omar, M.F.; Soong, R.; Benoukraf, T. Methodological aspects of whole-genome bisulfite sequencing analysis. Brief. Bioinform. 2014, 16, 369–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xi, Y.; Li, W. BSMAP: Whole genome bisulfite sequence MAPping program. BMC Bioinf. 2009, 10, 232. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, S.; Otto, C.; Kurtz, S.; Sharma, C.M.; Khaitovich, P.; Vogel, J.; Stadler, P.F.; Hackermüller, J. Fast mapping of short sequences with mismatches, insertions and deletions using index structures. PLoS Comput. Biol. 2009, 5, e1000502. [Google Scholar] [CrossRef] [PubMed]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Fiziev, P.; Yan, W.; Cokus, S.; Sun, X.; Zhang, M.Q.; Chen, P.Y.; Pellegrini, M. BS-Seeker2: A versatile aligning pipeline for bisulfite sequencing data. BMC Genom. 2013, 14, 774. [Google Scholar] [CrossRef] [PubMed]
- Rakyan, V.K.; Down, T.A.; Balding, D.J.; Beck, S. Epigenome-wide association studies for common human diseases. Nat. Rev. Genet. 2011, 12, 529. [Google Scholar] [CrossRef] [PubMed]
- Masser, D.R.; Berg, A.S.; Freeman, W.M. Focused, high accuracy 5-methylcytosine quantitation with base resolution by benchtop next-generation sequencing. Epigenet. Chromatin 2013, 6, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masser, D.R.; Stanford, D.R.; Freeman, W.M. Targeted DNA methylation analysis by next-generation sequencing. J. Vis. Exp. 2015. [Google Scholar] [CrossRef] [PubMed]
- Bashtrykov, P.; Jeltsch, A. DNA methylation analysis by bisulfite conversion coupled to double multiplexed amplicon-based next-generation sequencing (NGS). In Epigenome Editing; Springer Nature: Basel, Switzerland, 2018; pp. 367–382. [Google Scholar]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.Q.; Ali, I.; Tang, J.; Yang, W.C. New insights into 5hmC DNA modification: Generation, distribution and function. Front. Genet. 2017, 8, 100. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Pastor, W.A.; Shen, Y.; Tahiliani, M.; Liu, D.R.; Rao, A. The behaviour of 5-hydroxymethylcytosine in bisulfite sequencing. PLoS ONE 2010, 5, e8888. [Google Scholar] [CrossRef] [PubMed]
- Hadad, N.; Masser, D.R.; Logan, S.; Wronowski, B.; Mangold, C.A.; Clark, N.; Otalora, L.; Unnikrishnan, A.; Ford, M.M.; Giles, C.B.; et al. Absence of genomic hypomethylation or regulation of cytosine-modifying enzymes with aging in male and female mice. Epigenet. Chromatin 2016, 9, 30. [Google Scholar] [CrossRef] [PubMed]
- Booth, M.J.; Marsico, G.; Bachman, M.; Beraldi, D.; Balasubramanian, S. Quantitative sequencing of 5-formylcytosine in DNA at single-base resolution. Nat. Chem. 2014, 6, 435. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.G.; Gross, J.A.; Lutz, P.E.; Vaillancourt, K.; Maussion, G.; Bramoulle, A.; Théroux, J.F.; Gardini, E.S.; Ehlert, U.; Bourret, G.; et al. Medium throughput bisulfite sequencing for accurate detection of 5-methylcytosine and 5-hydroxymethylcytosine. BMC Genom. 2017, 18, 96. [Google Scholar] [CrossRef] [PubMed]
- Song, C.-X.; Yi, C.; He, C. Mapping recently identified nucleotide variants in the genome and transcriptome. Nat. Biotechnol. 2012, 30, 1107. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Hon, G.C.; Szulwach, K.E.; Song, C.X.; Jin, P.; Ren, B.; He, C. Tet-assisted bisulfite sequencing of 5-hydroxymethylcytosine. Nat. Protoc. 2012, 7, 2159. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Hon, G.C.; Szulwach, K.E.; Song, C.X.; Zhang, L.; Kim, A.; Li, X.; Dai, Q.; Shen, Y.; Park, B.; et al. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell 2012, 149, 1368–1380. [Google Scholar] [CrossRef] [PubMed]
- Booth, M.J.; Branco, M.R.; Ficz, G.; Oxley, D.; Krueger, F.; Reik, W.; Balasubramanian, S. Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science 2012, 336, 934–937. [Google Scholar] [CrossRef] [PubMed]
- Dapprich, J.; Ferriola, D.; Mackiewicz, K.; Clark, P.M.; Rappaport, E.; D’Arcy, M.; Sasson, A.; Gai, X.; Schug, J.; Kaestner, K.H.; et al. The next generation of target capture technologies-large DNA fragment enrichment and sequencing determines regional genomic variation of high complexity. BMC Genom. 2016, 17, 486. [Google Scholar] [CrossRef] [PubMed]
- Schmidl, C.; Klug, M.; Boeld, T.J.; Andreesen, R.; Hoffmann, P.; Edinger, M.; Rehli, M. Lineage-specific DNA methylation in T cells correlates with histone methylation and enhancer activity. Genome Res. 2009. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, R.A.; Ladd-Acosta, C.; Wen, B.; Wu, Z.; Montano, C.; Onyango, P.; Cui, H.; Gabo, K.; Rongione, M.; Webster, M.; et al. The human colon cancer methylome shows similar hypo-and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 2009, 41, 178. [Google Scholar] [CrossRef] [PubMed]
- Bäckdahl, L.; Herberth, M.; Wilson, G.; Tate, P.; Campos, L.S.; Cortese, R.; Eckhardt, F.; Beck, S. Gene body methylation of the dimethylarginine dimethylamino-hydrolase 2 (Ddah2) gene is an epigenetic biomarker for neural stem cell differentiation. Epigenetics 2009, 4, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Clark, T.A.; Spittle, K.E.; Turner, S.W.; Korlach, J. Direct detection and sequencing of damaged DNA bases. Genome Integr. 2011, 2, 10. [Google Scholar] [CrossRef] [PubMed]
- Bahassi, E.M.; Stambrook, P.J. Next-generation sequencing technologies: Breaking the sound barrier of human genetics. Mutagenesis 2014, 29, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Koren, S.; Miga, K.H.; Quick, J.; Rand, A.C.; Sasani, T.A.; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 2018, 36, 338. [Google Scholar] [CrossRef] [PubMed]
- Schatz, M.C. Nanopore sequencing meets epigenetics. Nat. Methods 2017, 14, 347. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; De Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Island, B.C. BRCA1 CpG island hypermethylation predicts sensitivity to poly(adenosine diphosphate)-ribose polymerase inhibitors. J. Clin. Oncol. 2010, 28, e563–e564. [Google Scholar]
- Berglund, E.C.; Kiialainen, A.; Syvänen, A.-C. Next-generation sequencing technologies and applications for human genetic history and forensics. Investig. Genet. 2011, 2, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reuter, J.A.; Spacek, D.V.; Snyder, M.P. High-throughput sequencing technologies. Mol. Cell 2015, 58, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, E.L.; Auger, H.; Jaszczyszyn, Y.; Thermes, C. Ten years of next-generation sequencing technology. Trends Genet. 2014, 30, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Eid, J.; Fehr, A.; Gray, J.; Luong, K.; Lyle, J.; Otto, G.; Peluso, P.; Rank, D.; Baybayan, P.; Bettman, B.; et al. Real-time DNA sequencing from single polymerase molecules. Science 2009, 323, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Veras, A.A.O.; de Sá, P.H.C.G.; Pinheiro, K.C.; Assis das Graças, D.; Azevedo Baraúna, R.; Cruz Schneider, M.P.; Azevedo, V.; Ramos, R.T.J.; Silva, A. Efficiency of Corynebacterium pseudotuberculosis Cp31 genome assembly with the Hi-Q enzyme on an Ion Torrent PGM sequencing platform. J. Proteom. Bioinform. 2014, 7, 374–378. [Google Scholar]
- Mardis, E.R. Next-generation sequencing platforms. Ann. Rev. Anal. Chem. 2013, 6, 287–303. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Fiddes, I.T.; Miga, K.H.; Olsen, H.E.; Paten, B.; Akeson, M. Improved data analysis for the MinION nanopore sequencer. Nat. Methods 2015, 12, 351. [Google Scholar] [CrossRef] [PubMed]
- Miles, B.N.; Ivanov, A.P.; Wilson, K.A.; Doğan, F.; Japrung, D.; Edel, J.B. Single molecule sensing with solid-state nanopores: Novel materials, methods, and applications. Chem. Soc. Rev. 2013, 42, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Van den Oord, E.J.; Bukszar, J.; Rudolf, G.; Nerella, S.; McClay, J.L.; Xie, L.Y.; Aberg, K.A. Estimation of CpG coverage in whole methylome next-generation sequencing studies. BMC Bioinform. 2013, 14, 50. [Google Scholar] [CrossRef] [PubMed]
- Hung, S.S.; Meissner, B.; Chavez, E.A.; Ben-Neriah, S.; Ennishi, D.; Jones, M.R.; Shulha, H.P.; Chan, F.C.; Boyle, M.; Kridel, R.; et al. Assessment of capture and amplicon-based approaches for the development of a targeted next-generation sequencing pipeline to personalize lymphoma management. J. Mol. Diagn. 2018, 20, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Sun, D.; Rodriguez, B.; Zhao, Q.; Sun, H.; Zhang, Y.; Li, W. BSeQC: Quality control of bisulfite sequencing experiments. Bioinformatics 2013, 29, 3227–3229. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Noviski, A.; Yu, X. MethyQA: A pipeline for bisulfite-treated methylation sequencing quality assessment. BMC Bioinform. 2013, 14, 259. [Google Scholar] [CrossRef] [PubMed]
- Mikeska, T.; Candiloro, I.L.; Dobrovic, A. The implications of heterogeneous DNA methylation for the accurate quantification of methylation. Epigenomics 2010, 2, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Macaulay, I.C.; Ponting, C.P.; Voet, T. Single-cell multiomics: Multiple measurements from single cells. Trends Genet. 2017, 33, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Smallwood, S.A.; Lee, H.J.; Angermueller, C.; Krueger, F.; Saadeh, H.; Peat, J.; Andrews, S.R.; Stegle, O.; Reik, W.; Kelsey, G. Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat. Methods 2014, 11, 817. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Huang, K.; An, Q.; Du, G.; Hu, G.; Xue, J.; Zhu, X.; Wang, C.Y.; Xue, Z.; Fan, G. Simultaneous profiling of transcriptome and DNA methylome from a single cell. Genome Biol. 2016, 17, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angermueller, C.; Clark, S.J.; Lee, H.J.; Macaulay, I.C.; Teng, M.J.; Hu, T.X.; Krueger, F.; Smallwood, S.A.; Ponting, C.P.; Voet, T.; et al. Parallel single-cell sequencing links transcriptional and epigenetic heterogeneity. Nat. Methods 2016, 13, 229. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Guo, H.; Cao, C.; Li, X.; Hu, B.; Zhu, P.; Wu, X.; Wen, L.; Tang, F.; Huang, Y.; et al. Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas. Cell Res. 2016, 26, 304. [Google Scholar] [CrossRef] [PubMed]
- Tanić, M.; Beck, S. Epigenome-wide association studies for cancer biomarker discovery in circulating cell-free DNA: Technical advances and challenges. Curr. Opin. Genet. Dev. 2017, 42, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Vaca-Paniagua, F.; Oliver, J.; da Costa, A.N.; Merle, P.; McKay, J.; Herceg, Z.; Holmila, R. Targeted deep DNA methylation analysis of circulating cell-free DNA in plasma using massively parallel semiconductor sequencing. Epigenomics 2015, 7, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Janku, F.; Huang, H.J.; Claes, B.; Falchook, G.S.; Fu, S.; Hong, D.; Ramzanali, N.M.; Nitti, G.; Cabrilo, G.; Tsimberidou, A.M.; et al. BRAF mutation testing in cell-free DNA from the plasma of patients with advanced cancers using a rapid, automated molecular diagnostics system. Mol. Cancer Ther. 2016, 15, 1397–1404. [Google Scholar] [CrossRef] [PubMed]
- Janku, F.; Huang, H.J.; Fujii, T.; Shelton, D.N.; Madwani, K.; Fu, S.; Tsimberidou, A.M.; Piha-Paul, S.A.; Wheler, J.J.; Zinner, R.G.; et al. Multiplex KRASG12/G13 mutation testing of unamplified cell-free DNA from the plasma of patients with advanced cancers using droplet digital polymerase chain reaction. Ann. Oncol. 2017, 28, 642–650. [Google Scholar] [PubMed]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.; Luo, H.; Krawczyk, M.; Wei, W.; Wang, W.; Wang, J.; Flagg, K.; Hou, J.; Zhang, H.; Yi, S.; et al. DNA methylation markers for diagnosis and prognosis of common cancers. Proc. Natl. Acad. Sci. USA 2017, 114, 7414–7419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janku, F.; Zhang, S.; Waters, J.; Liu, L.; Huang, H.J.; Subbiah, V.; Hong, D.S.; Karp, D.D.; Fu, S.; Cai, X.; et al. Development and validation of an ultradeep next-generation sequencing assay for testing of plasma cell-free DNA from patients with advanced cancer. Clin. Cancer Res. 2017, 23, 5648–5656. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, A.F.; Assenov, Y.; Martin-Subero, J.I.; Balint, B.; Siebert, R.; Taniguchi, H.; Yamamoto, H.; Hidalgo, M.; Tan, A.C.; Galm, O.; et al. A DNA methylation fingerprint of 1628 human samples. Genome Res. 2012, 22, 407–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moran, S.; Martínez-Cardús, A.; Sayols, S.; Musulén, E.; Balañá, C.; Estival-Gonzalez, A.; Moutinho, C.; Heyn, H.; Diaz-Lagares, A.; de Moura, M.C.; et al. Epigenetic profiling to classify cancer of unknown primary: A multicentre, retrospective analysis. Lancet Oncol. 2016, 17, 1386–1395. [Google Scholar] [CrossRef]
- Chan, K.A.; Jiang, P.; Zheng, Y.W.; Liao, G.J.; Sun, H.; Wong, J.; Siu, S.S.N.; Chan, W.C.; Chan, S.L.; Chan, A.T.; et al. Cancer genome scanning in plasma: Detection of tumor-associated copy number aberrations, single-nucleotide variants, and tumoral heterogeneity by massively parallel sequencing. Clin. Chem. 2013, 59, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Madic, J.; Kiialainen, A.; Bidard, F.C.; Birzele, F.; Ramey, G.; Leroy, Q.; Frio, T.R.; Vaucher, I.; Raynal, V.; Bernard, V.; et al. Circulating tumor DNA and circulating tumor cells in metastatic triple negative breast cancer patients. Int. J. Cancer 2015, 136, 2158–2165. [Google Scholar] [CrossRef] [PubMed]
- Couraud, S.; Paniagua, F.V.; Villar, S.; Oliver, J.; Schuster, T.; Blanche, H.; Girard, N.; Tredaniel, J.; Guilleminault, L.; Gervais, R.; et al. Non-invasive diagnosis of actionable mutations by deep sequencing of circulating-free DNA in non-small cell lung cancer: Findings from BioCAST/IFCT-1002. Clin. Cancer Res. 2014, 20, 4613–4624. [Google Scholar] [CrossRef] [PubMed]
- Warton, K.; Lin, V.; Navin, T.; Armstrong, N.J.; Kaplan, W.; Ying, K.; Gloss, B.; Mangs, H.; Nair, S.S.; Hacker, N.F.; et al. Methylation-capture and next-generation sequencing of free circulating DNA from human plasma. BMC Genom. 2014, 15, 476. [Google Scholar] [CrossRef] [PubMed]
- Lan, X.; Adams, C.; Landers, M.; Dudas, M.; Krissinger, D.; Marnellos, G.; Bonneville, R.; Xu, M.; Wang, J.; Huang, T.H.M.; et al. High resolution detection and analysis of CpG dinucleotides methylation using MBD-Seq technology. PLoS ONE 2011, 6, e22226. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.S.; Coolen, M.W.; Stirzaker, C.; Song, J.Z.; Statham, A.L.; Strbenac, D.; Robinson, M.D.; Clark, S.J. Comparison of methyl-DNA immunoprecipitation (MeDIP) and methyl-CpG binding domain (MBD) protein capture for genome-wide DNA methylation analysis reveal CpG sequence coverage bias. Epigenetics 2011, 6, 34–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Evolution of next-generation sequencing-based techniques applied to DNA methylation profiling. BS-Seq: bisulfite sequencing; MeDIP-Seq: methylated DNA immunoprecipitation sequencing; RRBS-Seq: reduced representation bisulfite sequencing; WGBS: whole genome bisulfite sequencing; MethylCap-Seq: methylation capture sequencing; MBD-Seq: methyl-CpG binding domain sequencing; oxBS.Seq: oxidative bisulfite sequencing; TAB-Seq: TET-associated bisulfite sequencing; BSAS: bisulfite amplicon sequencing.
Figure 1.
Evolution of next-generation sequencing-based techniques applied to DNA methylation profiling. BS-Seq: bisulfite sequencing; MeDIP-Seq: methylated DNA immunoprecipitation sequencing; RRBS-Seq: reduced representation bisulfite sequencing; WGBS: whole genome bisulfite sequencing; MethylCap-Seq: methylation capture sequencing; MBD-Seq: methyl-CpG binding domain sequencing; oxBS.Seq: oxidative bisulfite sequencing; TAB-Seq: TET-associated bisulfite sequencing; BSAS: bisulfite amplicon sequencing.


{kind=link}
{kind=link}
Table 1.
Comparison of key features of the different genome-wide approaches for DNA methylation profiling. CpGs: Cytosine-phosphate-Guanine; bp: base pair.
Table 1.
Comparison of key features of the different genome-wide approaches for DNA methylation profiling. CpGs: Cytosine-phosphate-Guanine; bp: base pair.
| Affinity Enrichment-Based Methods | Restriction Enzymes-Based Methods | Bisulfite Conversion-Based Methods | |
|---|---|---|---|
| Resolution | ~150 bp | Single-base | Single-base |
| Reads/sample | ~30–50 million reads | ~10 million reads | >500 million reads |
| CpGs covered | ~23 million CpGs | ~2 million CpGs | >28 million CpGs |
| Pros | Cost-effective method No mutations introduced | High sensitivity with lower costs | Evaluate methylation status of every CpG site |
| Cons | Biased toward hypermethylated regions Inability to predict absolute methylation level | CpGs in regions without the enzyme restriction site are not covered | Higher costs Requires high DNA input Substantial DNA degradation after bisulfite treatment |
| Application | Suitable for rapid, large scale and low-resolution studies | Suitable for site-specific/targeted studies | Suitable for high resolution studies |
Table 2.
Comparison of characteristics of the main next-generation sequencing technologies.
| Sequencing Platform Developers | Sequencing Principle | Key Features | Limitations | Reference |
|---|---|---|---|---|
| Illumina | Sequencing by synthesis | High throughput | Higher cost per read | [55,56,57] |
| Life Technologies Ion Torrent | Polymerization | Simple detection method | Low read number per run | [58,59] |
| Pacific Biosciences PacBio | Single molecule real time ligation | Single molecule detection and long read length | High error rates (13%) and low read number per run | [60] |
| Oxford Nanopore | Nanopore sensing | Single molecule and label-free detection with reduced costs | High error rates (38.2%) | [61,62] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Barros-Silva, D.; Marques, C.J.; Henrique, R.; Jerónimo, C. Profiling DNA Methylation Based on Next-Generation Sequencing Approaches: New Insights and Clinical Applications. Genes 2018, 9, 429. https://doi.org/10.3390/genes9090429
AMA Style
Barros-Silva D, Marques CJ, Henrique R, Jerónimo C. Profiling DNA Methylation Based on Next-Generation Sequencing Approaches: New Insights and Clinical Applications. Genes. 2018; 9(9):429. https://doi.org/10.3390/genes9090429
Chicago/Turabian StyleBarros-Silva, Daniela, C. Joana Marques, Rui Henrique, and Carmen Jerónimo. 2018. "Profiling DNA Methylation Based on Next-Generation Sequencing Approaches: New Insights and Clinical Applications" Genes 9, no. 9: 429. https://doi.org/10.3390/genes9090429
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.