RECTA: Regulon Identification Based on Comparative Genomics and Transcriptomics Analysis
by

Xin Chen
1,†, 
Anjun Ma
2,3,†,
Adam McDermaid
2,3,
Hanyuan Zhang
4,
Chao Liu
5,
Huansheng Cao
6 and
Qin Ma
2,3,* 1
Center for Applied Mathematics, Tianjin University, Tianjin 300072, China
2
Bioinformatics and Mathematical Biosciences Lab, Department of Agronomy, Horticulture and Plant Science, South Dakota State University, Brookings, SD 57006, USA
3
Department of Mathematics and Statistics, South Dakota State University, Brookings, SD 57006, USA
4
College of Computer Science and Engineering, University of Nebraska Lincoln, Lincoln, NE 68588, USA
5
Shandong Provincial Hospital affiliated to Shandong University, Jinan 250021, China
6
Center for Fundamental and Applied Microbiomics, Biodesign Institute, Arizona State University, Tempe, AZ 85287, USA
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Genes 2018, 9(6), 278; https://doi.org/10.3390/genes9060278
Submission received: 30 March 2018
/
Revised: 19 May 2018
/
Accepted: 25 May 2018
/
Published: 30 May 2018
(This article belongs to the Special Issue Selected Papers from the Bioinformatics and Intelligent Information Processing Conference (BIIP2018))
Abstract
:Regulons, which serve as co-regulated gene groups contributing to the transcriptional regulation of microbial genomes, have the potential to aid in understanding of underlying regulatory mechanisms. In this study, we designed a novel computational pipeline, regulon identification based on comparative genomics and transcriptomics analysis (RECTA), for regulon prediction related to the gene regulatory network under certain conditions. To demonstrate the effectiveness of this tool, we implemented RECTA on Lactococcus lactis MG1363 data to elucidate acid-response regulons. A total of 51 regulons were identified, 14 of which have computational-verified significance. Among these 14 regulons, five of them were computationally predicted to be connected with acid stress response. Validated by literature, 33 genes in Lactococcus lactis MG1363 were found to have orthologous genes which were associated with six regulons. An acid response related regulatory network was constructed, involving two trans-membrane proteins, eight regulons (llrA, llrC, hllA, ccpA, NHP6A, rcfB, regulons #8 and #39), nine functional modules, and 33 genes with orthologous genes known to be associated with acid stress. The predicted response pathways could serve as promising candidates for better acid tolerance engineering in Lactococcus lactis. Our RECTA pipeline provides an effective way to construct a reliable gene regulatory network through regulon elucidation, and has strong application power and can be effectively applied to other bacterial genomes where the elucidation of the transcriptional regulation network is needed.
1. Introduction
Genomic and transcriptomic analyses have been widely used for elucidating gene regulatory network (GRN) hierarchies and offering insight into the coordination of response capabilities in microorganisms [1,2,3,4]. One way to study the mechanism of transcriptional regulation in microbe genomics is regulon prediction. A regulon is a group of co-regulated operons, which contains single or multiple consecutive genes along the genome [5,6,7]. Genes in the same operon are controlled by the same promoter and are co-regulated by one or a set of transcriptional factors (TFs) [8]. The elucidation of regulons can improve the identification of transcriptional genes, and thus, reliably predict the gene transcription regulation networks [9].
There are three ways for regulon prediction: (i) predicting new operons for a known regulon [10,11]. This method combines motif profiling with a comparative genomic strategy to search for related regulon members and carries out systematical gene regulation study; (ii) Integrating cis-regulatory motif (motif for short) comparison and clustering to find significantly enriched motif candidates [12,13]. The candidate motifs are then assembled into regulons; (iii) Performing ab initio novel regulon inference using the de novo motif finding strategy [14]. This approach uses a phylogenetic footprinting technique which mostly relies on reference verification [15,16,17] and can perform a horizontal sequential comparison to predict regulons in target organisms by searching known functionally-related regulons or TFs from other relevant species. One algorithm for phylogenetic footprinting analysis called Motif Prediction by Phylogenetic footprinting (MP3) has been used for regulon prediction in Escherichia coli [17]. Phylogenetic footprinting was then integrated into the DMINDA webserver along with other algorithms, such as the Database of Prokaryotic Operons 2.0 (DOOR2) [7,18], Bottleneck Broken (BoBro) [19], and BoBro-based motif comparison (BBC) [13], to construct a complete pipeline for regulon prediction. In the latest research, a newly developed pipeline called Single-cell Regulatory Network Inference and Clustering (SCENIC) combines motif finding from co-expression gene modules (CEMs) with regulon prediction for single-cell clustering and analysis [20]. Such a method builds up a way of regulon application in single-cell and metagenomic research. Nevertheless, without a suitable regulon database, researchers need to build up the library first through operon identification, CEM analysis, motif prediction and comparison [21]. Here, we reported an integrated computational framework of regulon identification based on comparative genomics and transcriptomics analysis (RECTA) to elucidate the GRN responses in microbes under specific conditions. To better elucidate the methodology of RECTA, we built a regulatory network responding to the acid stress in Lactococcus lactis species.
Lactococcus lactis is one of the mesophilic Gram-positive lactic acid-producing bacteria. It has been widely applied in dairy fermentations, such as cheese and milk product [22]. Several studies have provided evidence of its essential roles in wrapping and delivering proteins or vaccinations for immune treatment, such as diabetes [23], malaria [24], tumors [25,26], and infections [27]. Holding the advantage of higher acid tolerance to protect vectors from resolving during delivery inside of the animal body, L. lactis has more potential and safety in oral drug development [28]. Moreover, it has been found that L. lactis, along with some Lactobacillus, Bifidobacterium, and other gut microbiota, were associated with obesity [29]. Such studies lead to the possibility and availability of L. lactis in metagenomic studies to investigate the effect of microbial interaction between L. lactis and other species in the human body. It is now well established that Lactococcus have evolved stress-sensing systems, which enable them to tolerate harsh environmental conditions [1,30,31].
Among the harsh environmental conditions that microorganisms confront, acid stress is known to change the level of the alarmones (guanosine tetraphosphate and guanosine pentaphosphate), collectively referred to as (p)ppGpp [32] and leads to a stringent response to cellular regulation [33]. The reason that bacteria maintain the protection mechanism against acid stress is to withstand the deleterious effects caused by the harmful high level of protons in the exposed environment. Many mechanisms or genes related to the acid stress response (ASR) have been identified. Proton-pumping activity, the direct regulator to acid stress response, controls the intracellular pH level by pumping extra protons out of the cell [34,35], and the increase of alkaline compound levels also counters the acidification found in Streptococcus [36]. Acid damage repair of cells by chaperones or proteases, such as GroES, GroEL, GrpE, HrcA, DnaK, DnaJ, Clp [37,38], hdeA/B and Hsp31 in E. coli [39,40], the arginine deiminase (ADI) system [41,42,43,44] and glutamate decarboxylases (GAD) pathways, and so on [45,46,47], have been proven to be associated with the acid response. Additionally, transcriptional regulators, σ factors, and two-component signal transduction system (TCSTs) have also been demonstrated to be responsible for ASR by modifying gene expression [48]. These genes or pathways suggest low pH has widespread adverse effects on cell functions and inflicts response at genomic, metabolic, and macromolecular levels. To better understand the mechanism that controls the acid tolerance and response to the acid stress in L. lactis, we considered MG1363, a strain extensively studied for acid resistance, to carry out computational analyses [1,49,50,51]. Nevertheless, to adequately describe the transcriptional state and gene regulation responsible for ASR in L. lactis, a GRN integrating all individual pathways is needed.
The experiment was conducted by six steps and the general framework is showcased in Figure 1: (i) MG1363 co-expression gene modules (CEMs) and differentially expressed genes (DEG) were generated from microarray data by hcluster package [52] and Wilcoxon test [53] in R, respectively. MG1363 operons were predicted from the genome sequence using the DOOR2 webserver and assigned into each CEM; (ii) for each CEM, the 300 bp upstream to the promoter was extracted and the sequences were used to find motifs using DMINDA 2.0; (iii) the top five significant motifs in each CEM were reassembled by their similarity comparison and clustering to predict regulons; (iv) the motifs were compared to known transcription factor binding sites (TFBSs) in the MEME suite [54], and the TFs corresponding to these TFBSs were mapped to MG1363 using basic local alignment search tool (BLAST). Only regulons with DEGs and mapped TF were kept as ASR-related regulons; (v) experimentally identified ASR-related genes in other organisms were mapped to MG1363 using BLAST and allocated to corresponding regulons for further verification; and (vi) the relationship between regulons and functional gene modules was established to elucidate the overall ASR mechanism in MG1363.
As a result, 14 regulons are identified, literature verified or putative, to be connected to ASR. Eight regulons, related to nine functional modules and 33 associated genes, are considered as the essential elements in acid resistance in MG1363. This proposed computational pipeline and the above results significantly expand the current understanding of the ASR system, providing a new method to predict systematic regulatory networks based on regulon clustering.
2. Materials and Methods
2.1. Data Acquisition
The L. lactic MG1363 genome sequence was downloaded from NCBI (GenBank accession number: AM406671). The microarray dataset containing eight samples under different acid stress conditions for MG1363 was downloaded from the Gene Expression Omnibus (GEO) database (Series number: GSE47012). The data has been treated with LOWESS normalization by the provider. The details on cell culture preparation and data processing can be found in the previous study [1]. This dataset has all bacteria grown in basic conditions: a two-liter fermenter in chemically defined medium containing 1% (w/v) glucose at 30 °C. The control and treatment samples were grown at a pH of 6.5 and 5.1, respectively.
Several TFBS databases integrated in the MEME suite, including DPInteract (E. coli) [55], JASPAR [56], RegTransBase (prokaryotes) [57], Prodoric Release (prokaryotes) [58], and Yeastract (yeast) [59], were utilized for regulon filtering in known TF templates to find homologous TFs and corresponding genes in MG1363 using BLAST with default parameters. In the literature validation part, all ASR-related transporters and genes were collected from published articles, and their sequences were obtained from NCBI and UniProt databases.
2.2. Operon Identification
The genome-scale operons of MG1363 were identified by DOOR2. It is a one-stop operon-centered resource including operons, alternative transcriptional units, motifs, terminators, and conserved operons information across multiple species [18]. Operons were predicted by the back-end prediction algorithm with a prediction accuracy of 90–95% [60], based on the features of intergenic distance, neighborhood conservation, short DNA motifs, length ratio between gene pairs, and newly developed transcriptomic features trained from the strand-specific RNA sequencing (RNA-Seq) dataset [61,62].
2.3. Gene Differential Expression Analysis and Co-Expression Analysis
Differentially expressed genes were identified based on the Wilcoxon signed-rank test [53] between the control and treatment, which was performed in R. The gene co-expression analysis was performed using a hierarchical clustering method (hcluster package in R) [52] to detect the CEMs under the acid stress in MG1363.
2.4. Motif finding and Regulon Prediction
Genes from each CEM were first mapped to the identified operons to retrieve the basic transcription units. Next, 300 bps in the upstream of the translation starting sites for each operon were extracted, in which motif finding was carried out using the webserver DMINDA [63,64], with the whole genome sequence used as the control set. DMINDA is a dominant motif prediction tool, embraced five analytical algorithms to find, scan, and compare motifs [13,61,65], including a phylogenetic footprint framework to elucidate the mechanism of transcriptional regulation at a system level in prokaryotic genomes [9,17,19]. A motif length of 12 nucleotides was used as the representative length for regulon prediction [12,13]. The sequences were uploaded to the server and default parameters were used in the BBC program to conduct motif clustering to find the top five significant motifs (p-value < 0.05) in each cluster. The identified motifs were subjected to motif comparison and grouped into regulons using Kruskal’s algorithm with two similarity thresholds, T1 and T2, to give rise to the highly reliable and relatively reliable motif clusters, respectively, in the BBC program in DMINDA [13].
2.5. Regulon Validation Based on Transcription Factor BLAST and Differentially Expressed Gene Filtering
Each highly conserved motif was considered to contain the same TFBS among species. Therefore, a comparison study was performed using Tomtom with default parameters in the MEME Suite [54] between identified motif and public-domain TFBS databases, including DPInteract, JASPAR, RegTransBase, Prodoric Release and Yeastract, to find TFBSs and corresponding TFs with significant p-values in other prokaryotic species. Those TFs were then mapped to MG1363 using BLAST by default parameters to predict the connection between regulons and TFs in MG1363. On the other hand, since genes without differential expression were supposed not to react to pH changes, and thus, irrelevant to ASR, regulons without DEGs were not involved in the GRN, and thus, excluded from the following steps.
2.6. Regulon Validation Based on Known Acid Stress Response Proteins from the Literature
To validate the performance of the above computational pipeline for regulon prediction, a literature-based validation was performed. Thirty-six ASR-related proteins and genes in other organisms including L. lactis, E. coli, Streptococcus, and so on were first manually collected from literature, and their sequence was retrieved from the NCBI and UniProt databases. They were used to examine the existing known mechanisms in response to pH changes in MG1363 using the BLAST program by default parameters on NCBI. Such literature-based validation can either confirm the putative regulons when known ASR-related genes can be found in the significant regulons or expand our results to some insufficiently significant regulons, which indicate both false positive and true negative rate to evaluate the computational pipeline.
3. Results
3.1. Predicted Operons and Co-Expression Gene Module Generation
A total of 1565 operons with 2439 coding genes of MG1363 (dataset S1) were retrieved from the DOOR2 database. Through co-expression analysis, the 1565 operons were grouped into 124 co-expressed clusters by calculating the Euclidean distance using h = 0.05 × (MAX (distance)). Among these clusters, two large groupings contain more than 200 operons. Each of which was removed from the subsequent analyses as larger clusters may have higher chances to induce false positive operons which were connected with true operons by co-expression analysis. For the remaining 122 clusters covering 2122 genes, 26 (21%) contain no more than 10 operons; the smallest cluster had two operons, and most of the clusters (90%) contained between 10 and 50 operons (dataset S2 and Figure S1).
3.2. Predicted Regulons Based on Motif Finding and Clustering
Using BoBro in the DMINDA webserver, multiple motif sequences were identified from the 300 bps in the upstream of the translation start sites for each operon. Only the top five significant motifs (adjusted p-value < 0.001) were selected in each cluster, giving rise to a total of 610 (122 × 5) identified. The motif comparison-and-clustering analysis was then performed on the 610 motifs, and 51 motif clusters were identified, with a motif similarity 0.8 as a cutoff. Intuitively, the operons sharing highly similar motifs in each motif cluster are supposed to be regulated by the same TF and tend to be in the same regulon. Hence, these 51 motif clusters correspond to 51 regulons (dataset S3).
3.3. Computationally-Verified Regulon Based on Transcription Factor BLAST and Differential Gene Expression Analysis
Among the above 51 regulons, 14 were found containing motifs significantly (E-value < 0.05) matched to known TFBSs using TOMTOM in the MEME suite, representatively. The motif logos are shown in Figure S2, and more details can be found in dataset S4. The 14 TFBS-corresponding TFs were then mapped to MG1363 using BLAST to identify the real TFs/genes regulating each regulon. As a result, eight known TFs—spo0A, lhfB, GAL80, CovR, c4494, ihfA, CovR, and RHE_PF00288—were successfully mapped to MG1363 resulting in eight TFs with multiple hits. The gene llrA (llmg_0908) regulates regulons #12 and #37, ccpA (llmg_0775) regulated regulons #15 and #47, hllA (llmg_0496) regulates regulons #7 and #31 (Table 1). The genes ccpA [66,67], llrA [68], llrC [68], and hllA [69], were known to be ASR-related genes in L. lactis; the gene llmg_0271, without any related known TF, was found to be similar to template TF GAL80 in yeast, which has not been associated with any ASR regulation pathways yet. For all 14 significant regulons, regulons #3, #4, #20, #28, #40, and #44 are potential candidates as, currently, no related TFs in L. lactis have been found (Table 1).
Additionally, 86 down-regulated genes and 55 up-regulated genes (dataset S5), resulting from DGE analysis were integrated into the regulons. Regulons #10, #37, #44 and #47 were found to be lacking DEGs. Thus, gene llmg_0271, related to regulon #10, was not likely to respond to acid stress in MG1363 even though it has been successfully mapped to MG1363, and was then grouped into the potential candidate. On the contrary, ccpA and llrA were still retained due to their involvements in regulons #15 and #12 with DEGs, respectively.
By the end of the computational pipeline, we predicted that regulons #2, #7, #12, #15 and #31 were related to GRN in MG1363 (Figure S3). A hypergeometric algorithm was used to verify the possibility of the of DEG numbers in each regulon (dataset S6). Merging regulon #7 and #31 as one, we referred to their TF names (ccpA, llrA, llrC, and hllA) to represent the five regulons for convenience.
3.4. Verified Regulons Based on Literature Verification
Altogether, 36 literature-supported ASR-related transporters were successfully mapped to MG1363 using blast with an E-value cutoff as 1e−10, which resulted in a total of 33 mapped genes. All the 36 transporters were categorized into nine modules based on their biological functions or regulated pathways, including L-lactate dehydrogenase (LDH), GAD, ADI, urea degradation, F1/F0ATPase, acid stress, protein repair and protease, envelope alterations, and DNA repair. The 33 mapped genes generate 22 operons and six regulons: llrA, llrC, hllA, NHP6A, regulon #8 and #39, which were subjected, one or more, to each functional module (Table 2).
Regulons llrA, llrC, and hllA have already been computationally identified in Table 1 and supported again by literature verification results. The NHP6A gene, interestingly, has a homologous TF in humans and fungi but not in L. lactis [70,71], yet failed to map in MG1363. Here, we are using NHP6A to represent regulon #20, as their relationship has been predicted computationally in Table 1. Regulon #39 was identified to be regulated by llrD, one of the six two-component regulatory systems in MG1363 [68]. Regulons #8 (llmg_1803) and #39 (llrD) were not included in the 14 significant regulons in Table 1. For NHP6A, regulons #8 and #39 were enriched by literature validation as it expanded regulon results of the RECTA pipeline. Among the nine functional modules, llrA was found connected to five of them, and NHP6A related to three. On the other hand, the GAD and urea degradation functional modules failed to connect to any previous regulons.
Compared to the regulon verification based on TF BLAST and DGE, the literature verification identified two more regulons (#8 and #39) that lay in the insignificant group, however, with no sign of ccpA regulon. Thus, such a result indicates a possible false positive rate of 1:5 and a true negative rate of 2:37 of our computational pipeline, indicating the reliability and feasibility of using RECTA to predict the ASR-related regulons. In Figure 2, we show the processes and results for both literature verification and the computational pipeline in detail. The final eight regulons predicted from both parts were then compared to construct a GRN response to acid stress, integrated with other information found in the literature.
3.5. A Model of Regulatory Network in Response to pH Change
According to the results outlined above, we are presenting a working model of the transcriptional regulatory network for acid stress response in MG1363 (Figure 3). The network consists of two transmembrane proteins (dataset S7), eight regulons, nine functional modules, and 33 orthologous genes known for ASR in other bacteria that are also contributing in MG1363.
The network is subjected to respond to the change of intracellular proton level. The signal is captured by H+ sensor and regulons are initiated to be regulated. Although significance was not shown for rcfB in our computational results, it has been reported to recognize and regulate promoter P170 [72], P1, and P3 [73,74], which are activated by boxes A, C and D (ACiD-box) and essential to acid response [75]. With the ACiD box, operons like groESL, lacticin 481 and lacZ have been proved to be regulated by rcfB, while als, aldB, etc., have not [75]. The homologous comparative study also predicted the existence of the ACiD box in llrA [68,76]. With such evidence, we separated rcfB from regulon #39 and predicted that rcfB is first triggered by H+ sensor and acts as the global initiator that controls the other seven regulons. It is reasonable that rcfB-related regulon #39 failed to show significant TF matching results after CEM treatment in the operon clustering step. The rcfB regulator worked as a trustworthy global factor; its differential expression should be less significant than regulons directly responding to acid stress, thus leading to the failure of being predicted by the RECTA pipeline. Nevertheless, the low number of microarray data sets (8) also limited the real performance to the ASR. However, the mechanism of how H+ sensor is activating and regulating the GRN and rcfB remains unclear. In the seven regulons, three—llrA, llrC and hllA—were verified through literature to be related to ASR; regulons #8 and #39 showed less significant in regulon prediction; NHP6A was considered as putative regulon due to its failure to map in MG1363; and ccpA was another putative regulon without literature support.
The six downstream regulons (llrA, llrC, hllA, NHP6A, regulon #39, and regulon #8) other than ccpA, interact with each other to regulate six ASR-related functional modules, including the ADI system, DNA repair, LDH, protein repair, envelope alterations, and F0/F1 adenosine triphosphatase (F0/F1ATPase). The ADI pathway, which generates adenosine triphosphate (ATP) and protects cells from acid stress [44], is under the regulation of NHP6A, llrC, llrA, and hllA. Another important pathway is the LDH (EC 1.1.1.27) under the regulation of NHP6A and llrA, which converts pyruvate and H+ to lactate which is exported outside of cells [77]. Chaperons which take part in macromolecule protection and repairing are subjected to regulon llrA. Chaperons have functions that include providing protection to against environmental stress, helping protein folding, and repairing damaged proteins, and have been demonstrated to show clear linkage with acid stress in numerous Gram-positive bacteria [37,38,39,40]. The F0/F1ATPase, controlled by llrA and regulon #8, also plays an important role in maintaining normal cellular pH, which pumps H+ out of cells at the expense of ATP [34,35,78,79]. The GAD [45,46] and urea degradation [48] functional modules are missing reliable associations with the regulons in MG1363 while maintaining functions in ASR mechanism in other species.
4. Discussion and Conclusions
Implementation of the novel computational pipeline RECTA resulted in the construction of an eight-regulons enrolled ASR regulatory network. The framework provides a useful tool and will be a starting point toward a more systems-level understanding of the question [80]. The identified motifs and regulons suggest acid resistance is a coordinated response regarding regulons, although most of these have not been identified or experimentally verified. From the three well-identified regulons—llrA, llrC, and hllA—it appears the gene regulation is also complex, as these regulons also interact with other proteins and TFs. The F0/F1ATPase is directly involved in the concentration regulations of the intracellular proton. Other pathways are responsible for repairing the damage caused by acid stress, such as DNA repair, protein repair, and cell envelops alterations. However, there were also several reported ASR-related genes or transporters such as htrA in Clostridium spp. [81], CovS/CovR acid response regulator in Streptococcus [82], cyclopropane fatty acid (cfa) synthase for cell-membrane modification [83], and oxidative damage protectant genes like sodA, nox-1 and nox-2 [84] that failed to map to MG1363. Using more gene expression datasets for CEM and DGE analyses could be a way to strengthen the result of our computational pipeline, which might cover more significant regulons to construct a more solid and complete regulatory network.
Homology mapping at the genomic level showed very a long evolutionary distance between MG1363 and currently well-annotated model species. Hence, the functional analysis for MG1363 is limited, and it is hard to apply gene functional enrichment to verify our prediction results. With more expression datasets and experiments about protein–protein interactions, the ASR mechanism can be largely improved in L. lactic MG1363.
In summary, through the implementation of RECTA, we found that the ASR at the transcriptome level in MG1363 is an orchestrated complex network. Functional annotation shows these regulons are involved in many levels of biological processes, including but not limited to DNA expression, transcription, and metabolism. Our method builds a TF-regulons-GRN relationship so that the new ASR-related genes can be predicted. Besides, the low false positive and true negative rate indicate the RECTA pipeline as sensitive and reasonable. In fact, considering the high accuracy, we regarded ccpA as the putative regulon, though not connected to any related functional modules, while more robust methods are required. Such results expand current pathways to those that can corroborate cell structures—cell wall, cell membranes, and so on—and related functions. Our findings suggest that acid has profound adverse effects and inflicts a systems-level response. Such predicted response pathways can inform better resistance design.
Looking forward to the acid tolerance advantage of L. lactis, which makes its prospective application in drug and vaccine delivery, the effects on anti-obesity research, and metagenomic studies, the ASR-related GRN in L. lactis shows an excellent research value. Fully understanding its theory may contribute to the development of Lactococcus therapy and can even expand to other close species by genetic modification. Furthermore, our computational pipeline provides an effective method to construct a reliable GRN based on regulon prediction, integrating CEMs, DGE analysis, motif finding, and comparative genomics study. It has a durable application power and can be effectively applied to other bacterial genomes, where the elucidation of the transcriptional regulation network is needed.
In this study, we designed a computational framework, RECTA, for acid-response regulon elucidation. This tool integrates differential gene expression, co-expression analysis, cis-regulatory motif identification, and comparative genomics to predict and validate regulons associated with acid response. In demonstrating the efficacy of this tool, we analyzed Lactococcus lactis MG1363. This implementation resulted in the expanded understanding of the acid-response regulon network for this one strain of L. lactis and provides an applicable method for acid-response regulon elucidation of further species. Through utilization of the RECTA pipeline, researchers can readily evaluate acid-response mechanisms for numerous bacterial species, while simultaneously validating the results of their study.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4425/9/6/278/s1. Figure S1: The distribution of operon numbers among 122 clusters, Figure S2: The motif logos extracted from 14 significant regulons, used for TFBS comparison. Figure S3: Heatmaps showing differential expression of the selected five regulons, Dataset S1: The operon prediction results for MG1363, Dataset S2: The gene clustering results for microarray GSE47012 containing operons. Dataset S3: The summarized motif prediction results for regulon prediction, Dataset S4: Computational validation results for predicted regulons, Dataset S5: 86 up-regulated genes and 55 down-regulated genes, Dataset S6: The possibility of the of DEG numbers in each regulon, Dataset S7: The prediction results for trans-membrane proteins.
Author Contributions
X.C. and Q.M. carried out the framework of the paper and are responsible for the regulon prediction work. H.Z. carried out the operon prediction and motif finding work. C.L. was responsible for DGE analysis. H.C., A.Ma and A.Mc. drafted and revised the manuscript. All authors submitted comments, read and approved the final manuscript.
Acknowledgments
This work was supported by National Science Foundation/EPSCoR Award No. IIA-1355423, the State of South Dakota Research Innovation Center and the Agriculture Experiment Station of South Dakota State University (SDSU). Support for this project was also provided by Hatch Project: SD00H558-15/project accession No. 1008151 from the USDA National Institute of Food and Agriculture, Sanford Health–SDSU Collaborative Research Seed Grant Program, and SDSU Scholarly Excellence Award (337T06). This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation (grant number ACI-1548562).
Conflicts of Interest
The authors declare that they have no competing interests.
Abbreviations
| ADI | Arginine deiminase |
| BBC | Bobro-based motif comparison |
| BoBro | Bottleneck broken |
| CEM | Co-expression (gene) module |
| DEG | Differentially expressed gene |
| DGE | Differential gene expression |
| E. coli | Escherichia coli |
| GAD | Glutamate decarboxylases |
| GRN | Gene regulatory network |
| LDH | Lactate dehydrogenase |
| L. lactis | Lactococcus lactis |
| MG1363 | Lactococcus lactis MG1363 |
| Motif | Cis-regulatory motif |
| RECTA | Regulon identification based on comparative genomics and transcriptomics analysis |
| TF | Transcription factors |
| TFBS | Transcriptional factor binding site |
References
- Carvalho, A.L.; Turner, D.L.; Fonseca, L.L.; Solopova, A.; Catarino, T.; Kuipers, O.P.; Voit, E.O.; Neves, A.R.; Santos, H. Metabolic and transcriptional analysis of acid stress in Lactococcus lactis, with a focus on the kinetics of lactic acid pools. PLoS ONE 2013, 8, e68470. [Google Scholar] [CrossRef] [PubMed]
- Locke, J.C.W.; Young, J.W.; Fontes, M.; Jiménez, M.J.H.; Elowitz, M.B. Stochastic pulse regulation in bacterial stress response. Science 2011, 334, 366–369. [Google Scholar] [CrossRef] [PubMed]
- Levine, J.H.; Lin, Y.; Elowitz, M.B. Functional roles of pulsing in genetic circuits. Science 2013, 342, 1193–1200. [Google Scholar] [CrossRef] [PubMed]
- Arnoldini, M.; Mostowy, R.; Bonhoeffer, S.; Ackermann, M. Evolution of stress response in the face of unreliable environmental signals. PLoS Comput. Biol. 2012, 8, e1002627. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Ma, Q.; Liu, B.; Chen, X.; Zhang, H.; Xu, Y. Revisiting operons: An analysis of the landscape of transcriptional units in E. coli. BMC Bioinform. 2015, 16, 356. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Ma, Q.; Li, G. Elucidation of operon structures across closely related bacterial genomes. PLoS ONE 2014, 9, e100999. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Ma, Q.; Chen, X.; Xu, Y. DOOR: A prokaryotic operon database for genome analyses and functional inference. Brief. Bioinform. 2017. [Google Scholar] [CrossRef] [PubMed]
- Jacob, F.; Perrin, D.; Sanchez, C.; Monod, J. Operon: A group of genes with the expression coordinated by an operator. C. R. Hebd. Seances Acad. Sci. 1960, 250, 1727–1729. [Google Scholar] [PubMed]
- Liu, B.; Zhou, C.; Li, G.; Zhang, H.; Zeng, E.; Liu, Q.; Ma, Q. Bacterial regulon modeling and prediction based on systematic cis regulatory motif analyses. Sci. Rep. 2016, 6, 23030. [Google Scholar] [CrossRef] [PubMed]
- Kumka, J.E.; Bauer, C.E. Analysis of the FnrL regulon in Rhodobacter capsulatus reveals limited regulon overlap with orthologues from Rhodobacter sphaeroides and Escherichia coli. BMC Genom. 2015, 16, 895. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.; Moreno-Hagelsieb, G.; Collado-Vides, J.; Stormo, G.D. A comparative genomics approach to prediction of new members of regulons. Genome Res. 2001, 11, 566–584. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Stamatoyannopoulos, J.A.; Bailey, T.L.; Noble, W.S. Quantifying similarity between motifs. Genome Biol. 2007, 8, R24. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Liu, B.; Zhou, C.; Yin, Y.; Li, G.; Xu, Y. An integrated toolkit for accurate prediction and analysis of cis-regulatory motifs at a genome scale. Bioinformatics 2013, 29, 2261–2268. [Google Scholar] [CrossRef] [PubMed]
- Novichkov, P.S.; Rodionov, D.A.; Stavrovskaya, E.D.; Novichkova, E.S.; Kazakov, A.E.; Gelfand, M.S.; Arkin, A.P.; Mironov, A.A.; Dubchak, I. RegPredict: An integrated system for regulon inference in prokaryotes by comparative genomics approach. Nucleic Acids Res. 2010, 38, W299–W307. [Google Scholar] [CrossRef] [PubMed]
- Blanchette, M.; Schwikowski, B.; Tompa, M. Algorithms for phylogenetic footprinting. J. Comput. Biol. 2002, 9, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Katara, P.; Grover, A.; Sharma, V. Phylogenetic footprinting: A boost for microbial regulatory genomics. Protoplasma 2012, 249, 901–907. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhang, H.; Zhou, C.; Li, G.; Fennell, A.; Wang, G.; Kang, Y.; Liu, Q.; Ma, Q. An integrative and applicable phylogenetic footprinting framework for cis-regulatory motifs identification in prokaryotic genomes. BMC Genom. 2016, 17, 578. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Ma, Q.; Zhou, C.; Chen, X.; Zhang, H.; Yang, J.; Mao, F.; Lai, W.; Xu, Y. DOOR 2.0: Presenting operons and their functions through dynamic and integrated views. Nucleic Acids Res. 2014, 42, D654–D659. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Ma, Q.; Mao, X.; Yin, Y.; Zhu, X.; Xu, Y. Integration of sequence-similarity and functional association information can overcome intrinsic problems in orthology mapping across bacterial genomes. Nucleic Acids Res. 2011, 39, e150. [Google Scholar] [CrossRef] [PubMed]
- Aibar, S.; Gonzalez-Blas, C.B.; Moerman, T.; Huynh-Thu, V.A.; Imrichova, H.; Hulselmans, G.; Rambow, F.; Marine, J.C.; Geurts, P.; Aerts, J.; et al. SCENIC: Single-cell regulatory network inference and clustering. Nat. Methods 2017, 14, 1083–1086. [Google Scholar] [CrossRef] [PubMed]
- Jensen, S.T.; Shen, L.; Liu, J.S. Combining phylogenetic motif discovery and motif clustering to predict co-regulated genes. Bioinformatics 2005, 21, 3832–3839. [Google Scholar] [CrossRef] [PubMed]
- Wegmann, U.; O’Connell-Motherway, M.; Zomer, A.; Buist, G.; Shearman, C.; Canchaya, C.; Ventura, M.; Goesmann, A.; Gasson, M.J.; Kuipers, O.P.; et al. Complete genome sequence of the prototype lactic acid bacterium Lactococcus lactis subsp. cremoris MG1363. J. Bacteriol. 2007, 189, 3256–3270. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Liu, J.; Hou, J.; Dong, Y.; Lu, Y.; Jin, L.; Cao, R.; Li, T.; Wu, J. Oral administration of recombinant Lactococcus lactis expressing HSP65 and tandemly repeated P277 reduces the incidence of type I diabetes in non-obese diabetic mice. PLoS ONE 2014, 9, e105701. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, R.; Yasawardena, S.; Zomer, A.; Venema, G.; Kok, J.; Leenhouts, K. Immunogenicity of a malaria parasite antigen displayed by Lactococcus lactis in oral immunisations. Vaccine 2006, 24, 3900–3908. [Google Scholar] [CrossRef] [PubMed]
- Bermudez-Humaran, L.G.; Cortes-Perez, N.G.; Lefevre, F.; Guimaraes, V.; Rabot, S.; Alcocer-Gonzalez, J.M.; Gratadoux, J.J.; Rodriguez-Padilla, C.; Tamez-Guerra, R.S.; Corthier, G.; et al. A novel mucosal vaccine based on live Lactococci expressing E7 antigen and IL-12 induces systemic and mucosal immune responses and protects mice against human papillomavirus type 16-induced tumors. J. Immunol. 2005, 175, 7297–7302. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Li, A.; Zuo, F.; Yu, R.; Zeng, Z.; Ma, H.; Chen, S. Recombinant Lactococcus lactis NZ9000 secretes a bioactive kisspeptin that inhibits proliferation and migration of human colon carcinoma HT-29 cells. Microb. Cell Fact. 2016, 15, 102. [Google Scholar] [CrossRef] [PubMed]
- Hanniffy, S.B.; Carter, A.T.; Hitchin, E.; Wells, J.M. Mucosal delivery of a pneumococcal vaccine using Lactococcus lactis affords protection against respiratory infection. J. Infect. Dis. 2007, 195, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Hols, P.; Kleerebezem, M.; Schanck, A.N.; Ferain, T.; Hugenholtz, J.; Delcour, J.; de Vos, W.M. Conversion of Lactococcus lactis from homolactic to homoalanine fermentation through metabolic engineering. Nat. Biotechnol. 1999, 17, 588–592. [Google Scholar] [CrossRef] [PubMed]
- Million, M.; Maraninchi, M.; Henry, M.; Armougom, F.; Richet, H.; Carrieri, P.; Valero, R.; Raccah, D.; Vialettes, B.; Raoult, D. Obesity-associated gut microbiota is enriched in Lactobacillus reuteri and depleted in Bifidobacterium animalis and Methanobrevibacter smithii. Int. J. Obes. 2012, 36, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Hutkins, R.W.; Nannen, N.L. pH homeostasis in lactic acid bacteria. J. Dairy Sci. 1993, 76, 2354–2365. [Google Scholar] [CrossRef]
- Van de Guchte, M.; Serror, P.; Chervaux, C.; Smokvina, T.; Ehrlich, S.D.; Maguin, E. Stress responses in lactic acid bacteria. Antonie Leeuwenhoek 2002, 82, 187–216. [Google Scholar] [CrossRef] [PubMed]
- Hauryliuk, V.; Atkinson, G.C.; Murakami, K.S.; Tenson, T.; Gerdes, K. Recent functional insights into the role of (p)ppGpp in bacterial physiology. Nat. Rev. Micro 2015, 13, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Rallu, F.; Gruss, A.; Ehrlich, S.D.; Maguin, E. Acid- and multistress-resistant mutants of Lactococcus lactis: Identification of intracellular stress signals. Mol. Microbiol. 2000, 35, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Koebmann, B.J.; Nilsson, D.; Kuipers, O.P.; Jensen, P.R. The membrane-bound H+-ATPase complex is essential for growth of Lactococcus lactis. J. Bacteriol. 2000, 182, 4738–4743. [Google Scholar] [CrossRef] [PubMed]
- Lund, P.; Tramonti, A.; De Biase, D. Coping with low pH: Molecular strategies in neutralophilic bacteria. FEMS Microbiol. Rev. 2014, 38, 1091–1125. [Google Scholar] [CrossRef] [PubMed]
- Shabayek, S.; Spellerberg, B. Acid Stress Response Mechanisms of Group B Streptococci. Front. Cell Infect. Microbiol. 2017, 7, 395. [Google Scholar] [CrossRef] [PubMed]
- Frees, D.; Vogensen, F.K.; Ingmer, H. Identification of proteins induced at low pH in Lactococcus lactis. Int. J. Food Microbiol. 2003, 87, 293–300. [Google Scholar] [CrossRef]
- Jayaraman, G.C.; Penders, J.E.; Burne, R.A. Transcriptional analysis of the Streptococcus mutans hrcA, grpE and dnaK genes and regulation of expression in response to heat shock and environmental acidification. Mol. Microbiol. 1997, 25, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Kern, R.; Malki, A.; Abdallah, J.; Tagourti, J.; Richarme, G. Escherichia coli HdeB is an acid stress chaperone. J. Bacteriol. 2007, 189, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Mujacic, M.; Baneyx, F. Chaperone Hsp31 contributes to acid resistance in stationary-phase Escherichia coli. Appl. Environ. Microbiol. 2007, 73, 1014–1018. [Google Scholar] [CrossRef] [PubMed]
- Budin-Verneuil, A.; Maguin, E.; Auffray, Y.; Ehrlich, D.S.; Pichereau, V. Genetic structure and transcriptional analysis of the arginine deiminase (ADI) cluster in Lactococcus lactis MG1363. Can. J. Microbiol. 2006, 52, 617–622. [Google Scholar] [CrossRef] [PubMed]
- Ryan, S.; Begley, M.; Gahan, C.G.; Hill, C. Molecular characterization of the arginine deiminase system in Listeria monocytogenes: Regulation and role in acid tolerance. Environ. Microbiol. 2009, 11, 432–445. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Fukamachi, T.; Saito, H.; Kobayashi, H. Adenosine deamination increases the survival under acidic conditions in Escherichia coli. J. Appl. Microbiol. 2012, 112, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Zuniga, M.; Perez, G.; Gonzalez-Candelas, F. Evolution of arginine deiminase (ADI) pathway genes. Mol. Phylogenet. Evol. 2002, 25, 429–444. [Google Scholar] [CrossRef]
- Nomura, M.; Nakajima, I.; Fujita, Y.; Kobayashi, M.; Kimoto, H.; Suzuki, I.; Aso, H. Lactococcus lactis contains only one glutamate decarboxylase gene. Microbiology 1999, 145, 1375–1380. [Google Scholar] [CrossRef] [PubMed]
- Sanders, J.W.; Leenhouts, K.; Burghoorn, J.; Brands, J.R.; Venema, G.; Kok, J. A chloride-inducible acid resistance mechanism in Lactococcus lactis and its regulation. Mol. Microbiol. 1998, 27, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Hoskins, J.; Alborn, W.E., Jr.; Arnold, J.; Blaszczak, L.C.; Burgett, S.; DeHoff, B.S.; Estrem, S.T.; Fritz, L.; Fu, D.J.; Fuller, W.; et al. Genome of the bacterium Streptococcus pneumoniae strain R6. J. Bacteriol. 2001, 183, 5709–5717. [Google Scholar] [CrossRef] [PubMed]
- Cotter, P.D.; Hill, C. Surviving the acid test: Responses of gram-positive bacteria to low pH. Microbiol. Mol. Biol. Rev. 2003, 67, 429–453. [Google Scholar] [CrossRef] [PubMed]
- Linares, D.M.; Kok, J.; Poolman, B. Genome sequences of Lactococcus lactis MG1363 (revised) and NZ9000 and comparative physiological studies. J. Bacteriol. 2010, 192, 5806–5812. [Google Scholar] [CrossRef] [PubMed]
- Sanders, J.W.; Venema, G.; Kok, J. Environmental stress responses in Lactococcus lactis. FEMS Microbiol. Rev. 1999, 23, 483–501. [Google Scholar] [CrossRef] [Green Version]
- Hartke, A.; Bouché, S.; Giard, J.C.; Benachour, A.; Boutibonnes, P.; Auffray, Y. The lactic acid stress response of Lactococcus lactis subsp. lactis. Curr. Microbiol. 1996, 33, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Antoine Lucas, S.J. Using AMAP and CTC Packages for Huge Clustering. R News 2006, 6, 58–60. [Google Scholar]
- Bauer, D.F. Constructing Confidence Sets Using Rank Statistics. J. Am. Stat. Assoc. 1972, 67, 687–690. [Google Scholar] [CrossRef]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME Suite: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef] [PubMed]
- DPInteract. Available online: http://arep.med.harvard.edu/dpinteract (accessed on 30 May 2018).
- Khan, A.; Fornes, O.; Stigliani, A.; Gheorghe, M.; Castro-Mondragon, J.A.; van der Lee, R.; Bessy, A.; Cheneby, J.; Kulkarni, S.R.; Tan, G.; et al. JASPAR 2018: Update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 2018, 46, D260–D266. [Google Scholar] [CrossRef] [PubMed]
- Kazakov, A.E.; Cipriano, M.J.; Novichkov, P.S.; Minovitsky, S.; Vinogradov, D.V.; Arkin, A.; Mironov, A.A.; Gelfand, M.S.; Dubchak, I. RegTransBase—A database of regulatory sequences and interactions in a wide range of prokaryotic genomes. Nucleic Acids Res. 2007, 35, D407–D412. [Google Scholar] [CrossRef] [PubMed]
- Munch, R.; Hiller, K.; Barg, H.; Heldt, D.; Linz, S.; Wingender, E.; Jahn, D. PRODORIC: Prokaryotic database of gene regulation. Nucleic Acids Res. 2003, 31, 266–269. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, M.C.; Monteiro, P.T.; Guerreiro, J.F.; Goncalves, J.P.; Mira, N.P.; dos Santos, S.C.; Cabrito, T.R.; Palma, M.; Costa, C.; Francisco, A.P.; et al. The YEASTRACT database: An upgraded information system for the analysis of gene and genomic transcription regulation in Saccharomyces cerevisiae. Nucleic Acids Res. 2014, 42, D161–D166. [Google Scholar] [CrossRef] [PubMed]
- Dam, P.; Olman, V.; Harris, K.; Su, Z.; Xu, Y. Operon prediction using both genome-specific and general genomic information. Nucleic Acids Res. 2007, 35, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Pachter, L. Models for transcript quantification from RNA-Seq. arXiv, 2011; arXiv:1104.3889. [Google Scholar]
- Chou, W.C.; Ma, Q.; Yang, S.; Cao, S.; Klingeman, D.M.; Brown, S.D.; Xu, Y. Analysis of strand-specific RNA-seq data using machine learning reveals the structures of transcription units in Clostridium thermocellum. Nucleic Acids Res. 2015, 43, e67. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Chen, X.; McDermaid, A.; Ma, Q. DMINDA 2.0: Integrated and systematic views of regulatory DNA motif identification and analyses. Bioinformatics 2017. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Zhang, H.; Mao, X.; Zhou, C.; Liu, B.; Chen, X.; Xu, Y. DMINDA: An integrated web server for DNA motif identification and analyses. Nucleic Acids Res. 2014. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Yang, J.; Li, Y.; McDermaid, A.; Ma, Q. An algorithmic perspective of de novo cis-regulatory motif finding based on ChIP-seq data. Brief. Bioinform. 2017. [Google Scholar] [CrossRef] [PubMed]
- Zomer, A.L.; Buist, G.; Larsen, R.; Kok, J.; Kuipers, O.P. Time-resolved determination of the CcpA regulon of Lactococcus lactis subsp. cremoris MG1363. J. Bacteriol. 2007, 189, 1366–1381. [Google Scholar] [CrossRef] [PubMed]
- Abranches, J.; Nascimento, M.M.; Zeng, L.; Browngardt, C.M.; Wen, Z.T.; Rivera, M.F.; Burne, R.A. CcpA regulates central metabolism and virulence gene expression in Streptococcus mutans. J. Bacteriol. 2008, 190, 2340–2349. [Google Scholar] [CrossRef] [PubMed]
- O’Connell-Motherway, M.; van Sinderen, D.; Morel-Deville, F.; Fitzgerald, G.F.; Ehrlich, S.D.; Morel, P. Six putative two-component regulatory systems isolated from Lactococcus lactis subsp. cremoris MG1363. Microbiology 2000, 146, 935–947. [Google Scholar] [CrossRef] [PubMed]
- Bolotin, A.; Mauger, S.; Malarme, K.; Ehrlich, S.D.; Sorokin, A. Low-redundancy sequencing of the entire Lactococcus lactis IL1403 genome. Antonie Leeuwenhoek 1999, 76, 27–76. [Google Scholar] [CrossRef] [PubMed]
- Kolodrubetz, D.; Burgum, A. Duplicated NHP6 genes of Saccharomyces cerevisiae encode proteins homologous to bovine high mobility group protein 1. J. Biol. Chem. 1990, 265, 3234–3239. [Google Scholar] [PubMed]
- Stillman, D.J. Nhp6: A small but powerful effector of chromatin structure in Saccharomyces cerevisiae. Biochim. Biophys. Acta 2010, 1799, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Madsen, S.M.; Arnau, J.; Vrang, A.; Givskov, M.; Israelsen, H. Molecular characterization of the pH-inducible and growth phase-dependent promoter P170 of Lactococcus lactis. Mol. Microbiol. 1999, 32, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Rince, A.; Dufour, A.; Le Pogam, S.; Thuault, D.; Bourgeois, C.M.; Le Pennec, J.P. Cloning, expression, and nucleotide sequence of genes involved in production of lactococcin DR, a bacteriocin from Lactococcus lactis subsp. lactis. Appl. Environ. Microbiol. 1994, 60, 1652–1657. [Google Scholar] [PubMed]
- Hindre, T.; Le Pennec, J.P.; Haras, D.; Dufour, A. Regulation of lantibiotic lacticin 481 production at the transcriptional level by acid pH. FEMS Microbiol. Lett. 2004, 231, 291–298. [Google Scholar] [CrossRef]
- Madsen, S.M.; Hindre, T.; Le Pennec, J.P.; Israelsen, H.; Dufour, A. Two acid-inducible promoters from Lactococcus lactis require the cis-acting ACiD-box and the transcription regulator RcfB. Mol. Microbiol. 2005, 56, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Akyol, I.; Comlekcioglu, U.; Karakas, A.; Serdaroglu, K.; Ekinci, M.S.; Ozkose, E. Regulation of the acid inducible rcfB promoter in Lactococcus lactis subsp. lactis. Ann. Microbiol. 2008, 58, 269. [Google Scholar] [CrossRef]
- Dennis, D.; Kaplan, N.O. d- and l-lactic acid dehydrogenases in Lactobacillus plantarum. J. Biol. Chem. 1960, 235, 810–818. [Google Scholar] [PubMed]
- Amachi, S.; Ishikawa, K.; Toyoda, S.; Kagawa, Y.; Yokota, A.; Tomita, F. Characterization of a mutant of Lactococcus lactis with reduced membrane-bound ATPase activity under acidic conditions. Biosci. Biotechnol. Biochem. 1998, 62, 1574–1580. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, E.; Condon, S. Relationship between acid tolerance, cytoplasmic pH, and ATP and H+-ATPase levels in chemostat cultures of Lactococcus lactis. Appl. Environ. Microbiol. 1999, 65, 2287–2293. [Google Scholar] [PubMed]
- Cao, H.; Wei, D.; Yang, Y.; Shang, Y.; Li, G.; Zhou, Y.; Ma, Q.; Xu, Y. Systems-level understanding of ethanol-induced stresses and adaptation in E. coli. Sci. Rep. 2017, 7, 44150. [Google Scholar] [CrossRef] [PubMed]
- Alsaker, K.V.; Paredes, C.; Papoutsakis, E.T. Metabolite stress and tolerance in the production of biofuels and chemicals: Gene-expression-based systems analysis of butanol, butyrate, and acetate stresses in the anaerobe Clostridium acetobutylicum. Biotechnol. Bioeng. 2010, 105, 1131–1147. [Google Scholar] [CrossRef] [PubMed]
- Cumley, N.J.; Smith, L.M.; Anthony, M.; May, R.C. The CovS/CovR acid response regulator is required for intracellular survival of group B Streptococcus in macrophages. Infect. Immun. 2012, 80, 1650–1661. [Google Scholar] [CrossRef] [PubMed]
- Budin-Verneuil, A.; Maguin, E.; Auffray, Y.; Ehrlich, S.D.; Pichereau, V. Transcriptional analysis of the cyclopropane fatty acid synthase gene of Lactococcus lactis MG1363 at low pH. FEMS Microbiol. Lett. 2005, 250, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Santi, I.; Grifantini, R.; Jiang, S.M.; Brettoni, C.; Grandi, G.; Wessels, M.R.; Soriani, M. CsrRS regulates group B Streptococcus virulence gene expression in response to environmental pH: A new perspective on vaccine development. J. Bacteriol. 2009, 191, 5387–5397. [Google Scholar] [CrossRef] [PubMed]
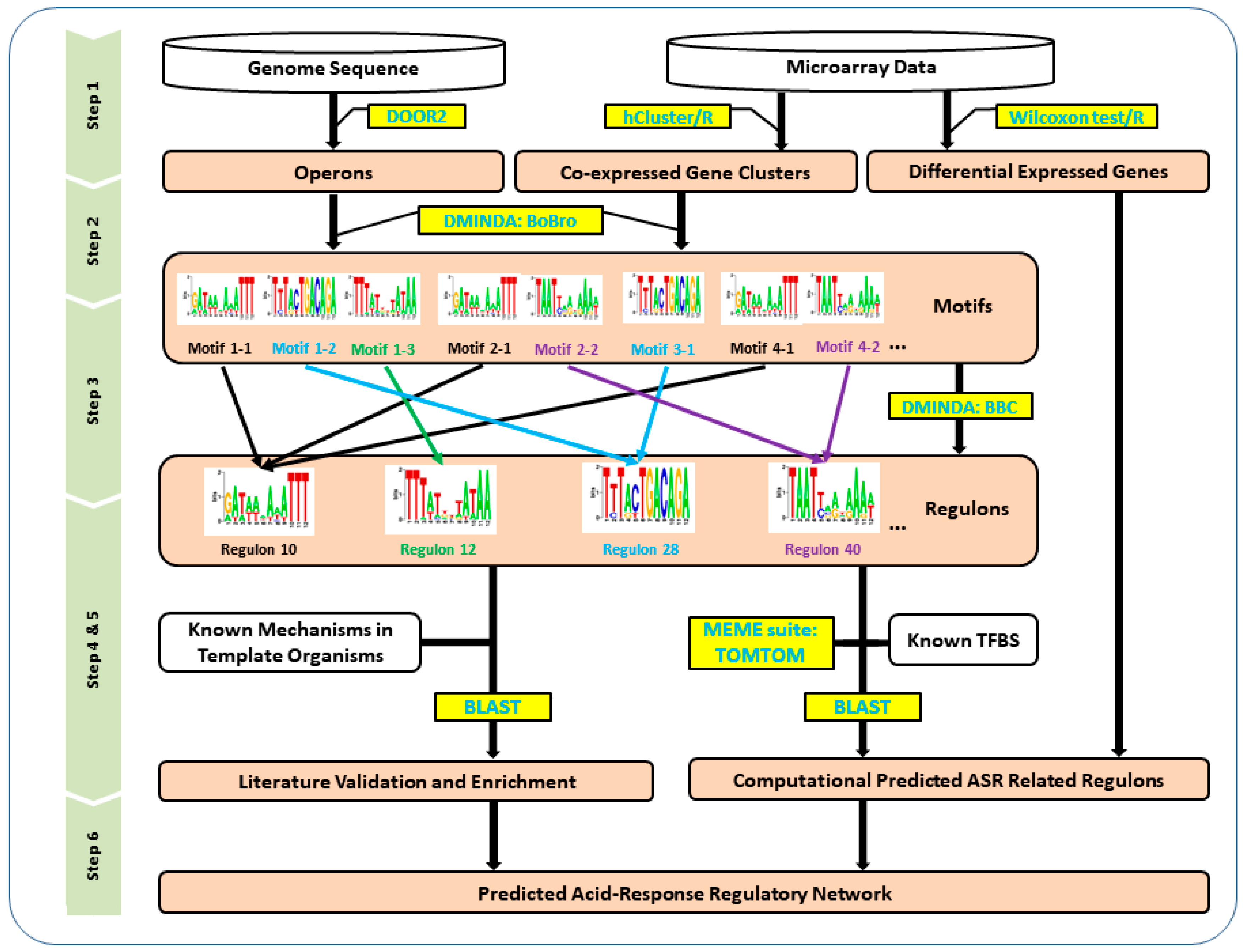
Figure 1.
The flowchart of constructing the global acid stress response (ASR) transcriptional network in MG1363. Step 1: microarray data was used to generate co-expressed gene clusters and differentially expressed genes (DEGs), and the MG1363 genome sequence was used to find operons. Step 2: a motif finding progress was carried out to identify all statistically significant motifs in each of the co-expression gene modules (CEMs). Step 3: a regulon finding procedure was designed to identify all the possible regulon candidates encoded in the genome based on motif comparison and clustering. Step 4: the motifs of each of these regulons were compared to known transcription factor binding sites (TFBSs), and differential gene expression (DGE) analysis between low pH conditions and normal conditions was used to figure out the ASR-related regulons. Step 5: regulon validation based on literature information verified the significant putative regulons and expanded the results to some insufficiently significant regulons. Step 6: the ASR-related gene regulatory network (GRN) in MG1363 was predicted and described with eight regulons, nine functional modules, and 33 genes. The combination of the above information forms a genome-scale regulatory network constructed for ASR. Abbreviations: DOOR2, Database of Prokaryotic Operons 2.0; BBC, BoBro-based motif comparison; BLAST, basic local alignment search tool; BoBro, Bottleneck Broken.
Figure 1.
The flowchart of constructing the global acid stress response (ASR) transcriptional network in MG1363. Step 1: microarray data was used to generate co-expressed gene clusters and differentially expressed genes (DEGs), and the MG1363 genome sequence was used to find operons. Step 2: a motif finding progress was carried out to identify all statistically significant motifs in each of the co-expression gene modules (CEMs). Step 3: a regulon finding procedure was designed to identify all the possible regulon candidates encoded in the genome based on motif comparison and clustering. Step 4: the motifs of each of these regulons were compared to known transcription factor binding sites (TFBSs), and differential gene expression (DGE) analysis between low pH conditions and normal conditions was used to figure out the ASR-related regulons. Step 5: regulon validation based on literature information verified the significant putative regulons and expanded the results to some insufficiently significant regulons. Step 6: the ASR-related gene regulatory network (GRN) in MG1363 was predicted and described with eight regulons, nine functional modules, and 33 genes. The combination of the above information forms a genome-scale regulatory network constructed for ASR. Abbreviations: DOOR2, Database of Prokaryotic Operons 2.0; BBC, BoBro-based motif comparison; BLAST, basic local alignment search tool; BoBro, Bottleneck Broken.

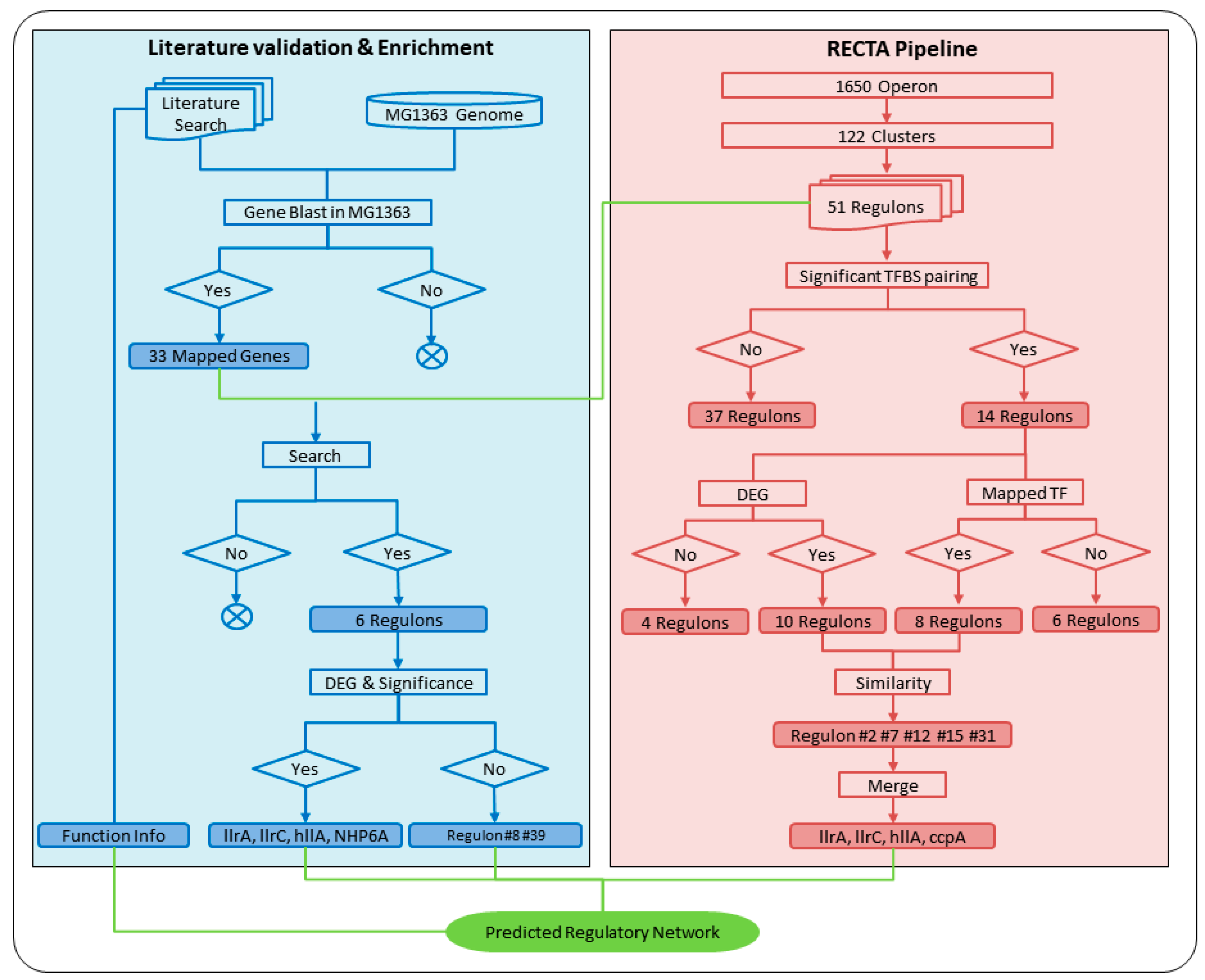
Figure 2.
Regulon prediction using regulon identification based on comparative genomics and transcriptomics analysis (RECTA) pipeline (red) and validation and enrichment using literature information and gene blast (blue). All processes were shown in rectangles and results were highlighted with corresponding background colors. In the computational pipeline, 51 regulons with assigned motifs and operons were analyzed sequentially through significant TFBS pairing, DEG conformation, and TF BLAST. Only regulons contained DEGs (10) which had related mapped TF (8) were believed to be the final predicted ASR-related regulons (5). These five regulons were then merged into four, using the corresponding TFs to represent their names. In the literature validation process, known ASR-related transporters were first mapped to the MG1363 genome and resulted in 33 genes. Those genes were then searched in 51 regulons and determined six related regulons. All regulons resulting from both computational pipeline and literature validation were combined, along with the information of functional modules, to determine the GRN.
Figure 2.
Regulon prediction using regulon identification based on comparative genomics and transcriptomics analysis (RECTA) pipeline (red) and validation and enrichment using literature information and gene blast (blue). All processes were shown in rectangles and results were highlighted with corresponding background colors. In the computational pipeline, 51 regulons with assigned motifs and operons were analyzed sequentially through significant TFBS pairing, DEG conformation, and TF BLAST. Only regulons contained DEGs (10) which had related mapped TF (8) were believed to be the final predicted ASR-related regulons (5). These five regulons were then merged into four, using the corresponding TFs to represent their names. In the literature validation process, known ASR-related transporters were first mapped to the MG1363 genome and resulted in 33 genes. Those genes were then searched in 51 regulons and determined six related regulons. All regulons resulting from both computational pipeline and literature validation were combined, along with the information of functional modules, to determine the GRN.

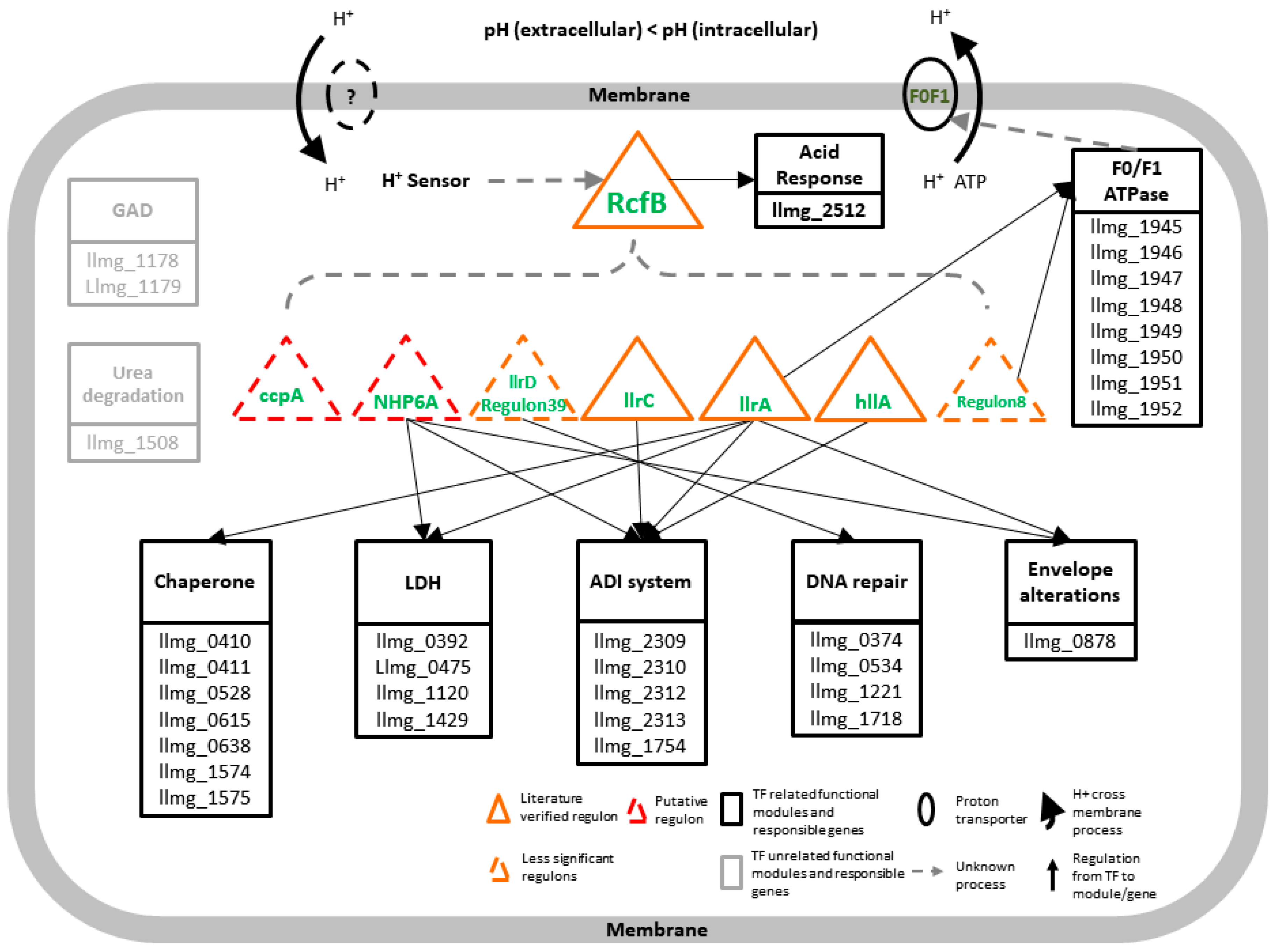
Figure 3.
A working model of the transcriptional gene regulatory network in response to pH change in L. lactis. The mechanism is activated by the change of proton signal in a cell. Regulon RcfB is assumed to be the overall activator for the rest seven regulons and controls the ASR functional module solely. Three kinds of literature were verified; significant ASR-related regulons, llrA, llrC, and hllA, and two insufficiently significant regulons, llrD (regulon #39) and regulon #8 (llmg_1803) were predicted via our workflow but with results under a 0.8 motif similarity cutoff or a hit could not be found; one putative significant regulon NHP6A controls the seven functional modules which are experimentally verified in the close species MG1363. The other significant regulon ccpA failed to be confirmed by any literature-proved genes or transporters. Two extra functional modules, GAD, and urea degradation show no direct connection to all seven of the regulons. One or more homology genes are found in MG1363 for all the nine modules using BLAST. The solid arrows indicate regulation between regulons/TFs and functional modules/genes, and the dashed arrows indicate uncertain control processes. Additionally, two ovals indicate two trans-membrane proteins; one is confirmed as F0/F1ATPase and the other one, with the dashed line, whose related information we cannot find in the public-domain literature.
Figure 3.
A working model of the transcriptional gene regulatory network in response to pH change in L. lactis. The mechanism is activated by the change of proton signal in a cell. Regulon RcfB is assumed to be the overall activator for the rest seven regulons and controls the ASR functional module solely. Three kinds of literature were verified; significant ASR-related regulons, llrA, llrC, and hllA, and two insufficiently significant regulons, llrD (regulon #39) and regulon #8 (llmg_1803) were predicted via our workflow but with results under a 0.8 motif similarity cutoff or a hit could not be found; one putative significant regulon NHP6A controls the seven functional modules which are experimentally verified in the close species MG1363. The other significant regulon ccpA failed to be confirmed by any literature-proved genes or transporters. Two extra functional modules, GAD, and urea degradation show no direct connection to all seven of the regulons. One or more homology genes are found in MG1363 for all the nine modules using BLAST. The solid arrows indicate regulation between regulons/TFs and functional modules/genes, and the dashed arrows indicate uncertain control processes. Additionally, two ovals indicate two trans-membrane proteins; one is confirmed as F0/F1ATPase and the other one, with the dashed line, whose related information we cannot find in the public-domain literature.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Altogether, 14 significant regulons that are verified and mapped to known transcription factors (TFs). According to analyses, operon numbers and DEG determination (yes or no), matched template TFs and mapped TFs were assigned for each significant regulon, respectively, and were aligned based on regulon ID number. Five regulons containing DEGs and having the corresponding TF at the same time were bolded, being computationally verified as the regulons responsible for acid stress in MG1363.
Table 1.
Altogether, 14 significant regulons that are verified and mapped to known transcription factors (TFs). According to analyses, operon numbers and DEG determination (yes or no), matched template TFs and mapped TFs were assigned for each significant regulon, respectively, and were aligned based on regulon ID number. Five regulons containing DEGs and having the corresponding TF at the same time were bolded, being computationally verified as the regulons responsible for acid stress in MG1363.
| Regulon ID | No. of Operons | DEG | TF Template | TF (Gene) BLAST in MG1363 |
|---|---|---|---|---|
| Regulon #2 | 82 | Y | spo0A | llrC (llmg_0414) |
| Regulon #3 | 32 | Y | FoxQ1 | N/A |
| Regulon #4 | 20 | Y | SPT2 | N/A |
| Regulon #7 | 49 | Y | lhfB | hllA (llmg_0496) |
| Regulon #10 | 5 | N | GAL80 | llmg_0271 |
| Regulon #12 | 259 | Y | CovR | llrA (llmg_0908) |
| Regulon #15 | 19 | Y | c4494 | ccpA (llmg_0775) |
| Regulon #20 | 79 | Y | NHP6A | N/A |
| Regulon #28 | 5 | Y | 1Z916 | N/A |
| Regulon #31 | 65 | Y | ihfA | hllA (llmg_0496) |
| Regulon #37 | 10 | N | CovR | llrA (llmg_0908) |
| Regulon #40 | 7 | Y | Awh | N/A |
| Regulon #44 | 12 | N | YBR182C | N/A |
| Regulon #47 | 5 | N | RHE_PF00288 | ccpA (llmg_0775) |
Abbreviations: N, no; Y, yes; N/A, not found.
Table 2.
Known ASR-related gene mapping from literature in response to pH change. Literature-supported ASR-related genes found in close species or other Lactococcus lactis strains. The template transporters and genes were first identified in published studies from the NCBI and UniProt databases. Lactococcus lactis Il1403 was used as the organism which is very close to MG1363 if template gene existed. Only 36 templates that successfully mapped to the MG1363 genome were listed, which resulted in 33 genes. All mapped genes and corresponding templated were organized by their regulated pathways which were further used as functional modules. Mapped genes were searched in 51 regulons to build the connections between functional modules and regulons.
Table 2.
Known ASR-related gene mapping from literature in response to pH change. Literature-supported ASR-related genes found in close species or other Lactococcus lactis strains. The template transporters and genes were first identified in published studies from the NCBI and UniProt databases. Lactococcus lactis Il1403 was used as the organism which is very close to MG1363 if template gene existed. Only 36 templates that successfully mapped to the MG1363 genome were listed, which resulted in 33 genes. All mapped genes and corresponding templated were organized by their regulated pathways which were further used as functional modules. Mapped genes were searched in 51 regulons to build the connections between functional modules and regulons.
| Template Organisms | MG1363 | |||
|---|---|---|---|---|
| Organisms | Transporters | Functions/Pathways | Mapped Genes (Locus Tag) | Regulons |
| Lactococcus lactis | ldh | LDH | ldh (llmg_1120) | NHP6A, llrA |
| ldhB | ldhB (llmg_0392, llmg_0475) | |||
| ldhX | ldhX (llmg_1429) | |||
| Lactococcus lactis | gadB | GAD | gadB (llmg_1179) | N/A |
| gadC | gadC (llmg_1178) | |||
| L actococcus lactis | arcA | ADI pathway | arcA (llmg_2313) | NHP6A, llrA, llrC, hllA |
| arcB | arcB (llmg_2312) | |||
| arcC1 | arcC1 (llmg_2310) | |||
| arcC2 | arcC2 (llmg_2309) | |||
| argF | argF (llmg_1754) | |||
| Bacteria | ureA/B/C$ | Urea degradation | pyrC (llmg_1508) | N/A |
| L actococcus lactis | atpEBFHAGDC$$ | F0/F1ATPase | llmg_1952, llmg_1951, llmg_1950, llmg_1949, llmg_1948, llmg_1947, llmg_1946, llmg_1945 | llrA, (Regulon8, llmg_1803) $$$ |
| Lactococcus lactis | rcfB | Acid response | rcfB (llmg_2512) | (Regulon39, llrD) $$$ |
| Lactococcus lactis, Escherichia coli K12 | dnak | Chaperone, Protein repair and protease | dnaK (llmg_1574) | llrA |
| groEL | groEL2 (llmg_0411) | |||
| groES | groES (llmg_0410) | |||
| grpE | grpE (llmg_1575) | |||
| clpE | clpE (llmg_0528) | |||
| clpC | clpC (llmg_0615) | |||
| clpP | clpP (llmg_0638) | |||
| Lactococcus lactis, Bacillus subtilis | dltC, agK, SGP, ffh | Envelope alterations | llmg_0878 | NHP6A, llrA |
| Lactococcus lactis | recA, uvr, smn | DNA repair | llmg_0374, llmg_0534, llmg_1718 llmg_1221 | (Regulon39, llrD) $$$ |
$ Three subunits of urease enzymes coded by ureABC operon found preserved in multiple bacteria. $$ Altogether eight genes. $$$ The homolog prediction or motif research results with low homolog similarity but have meaningful biological relevance. Abbreviations: LDH, L-lactate dehydrogenase; GAD, glutamate decarboxylases; ADI, arginine deiminase; N/A, not found.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Chen, X.; Ma, A.; McDermaid, A.; Zhang, H.; Liu, C.; Cao, H.; Ma, Q. RECTA: Regulon Identification Based on Comparative Genomics and Transcriptomics Analysis. Genes 2018, 9, 278. https://doi.org/10.3390/genes9060278
AMA Style
Chen X, Ma A, McDermaid A, Zhang H, Liu C, Cao H, Ma Q. RECTA: Regulon Identification Based on Comparative Genomics and Transcriptomics Analysis. Genes. 2018; 9(6):278. https://doi.org/10.3390/genes9060278
Chicago/Turabian StyleChen, Xin, Anjun Ma, Adam McDermaid, Hanyuan Zhang, Chao Liu, Huansheng Cao, and Qin Ma. 2018. "RECTA: Regulon Identification Based on Comparative Genomics and Transcriptomics Analysis" Genes 9, no. 6: 278. https://doi.org/10.3390/genes9060278
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.