
COBRA-Seq: Sensitive and Quantitative Methylome Profiling

Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Genomic DNA Isolation
2.2. Annealing Oligonucleotides to Construct COBRA-Seq Adapters

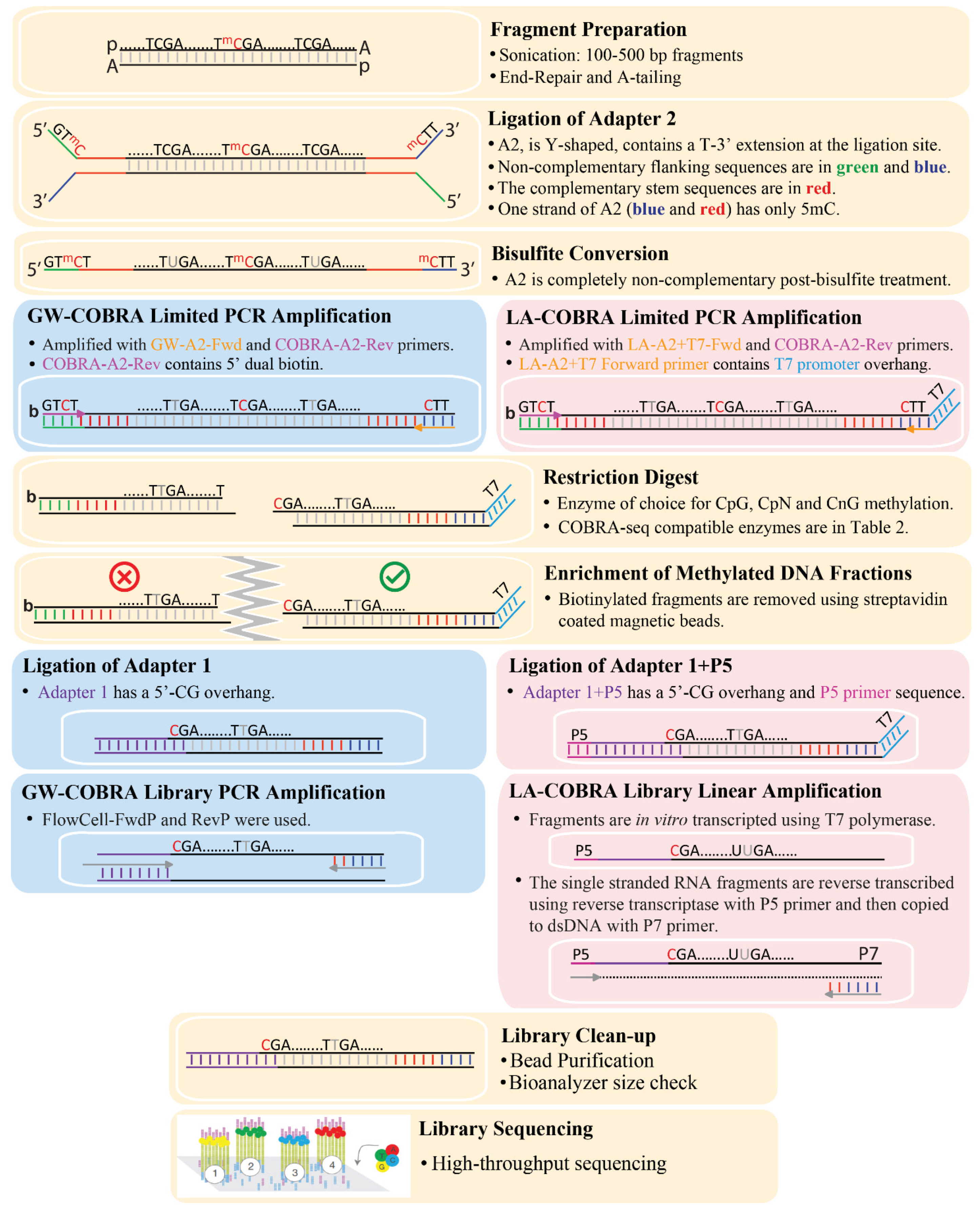{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Primer Sequence (5'–3')* | Purification Method |
|---|---|---|
| A2-UpperStrand | PAGE and HPLC | |
| A2-LowerStrand | PAGE and HPLC | |
| GW-A1-UpperStrand | CTA CAC TCT TTC CCT ACA CGA CGC TCT TCC GAT CT | PAGE and HPLC |
| GW-A1-LowerStrand | CGA GAT CGG AAG AGC GTC GTG TAG GGA AAG AGT GTA G | PAGE and HPLC |
| LA-A1+P5-UpperStrand | PAGE and HPLC | |
| LA-A1+P5-LowerStrand | PAGE and HPLC | |
| GW-A2-FwdP | CAA GCA GAA GAC GGC ATA CGA GCT CTT CCG ATC T | PAGE |
| COBRA-A2-RevP | b | HPLC |
| LA-A2+T7-FwdP | PAGE | |
| FlowCell-FwdP | AAT GAT ACG GCG ACC ACC GAG ATC TAC ACT CTT TCC CTA CAC GAC GCT CTT CCG ATC T | HPLC |
| FlowCell-RevP | CAA GCA GAA GAC GGC ATA CGA GCT CTT CCG ATC T | HPLC |
| LADS P5 | AAT GAT ACG GCG ACC ACC GA | HPLC |
| LADS P7 | CAA GCA GAA GAC GGC ATA CGA | HPLC |
2.3. Library Construction and Bisulfite Treatment
2.4. Bioinformatics and Statistics
3. Results and Discussion
3.1. COBRA-Seq Library Construction


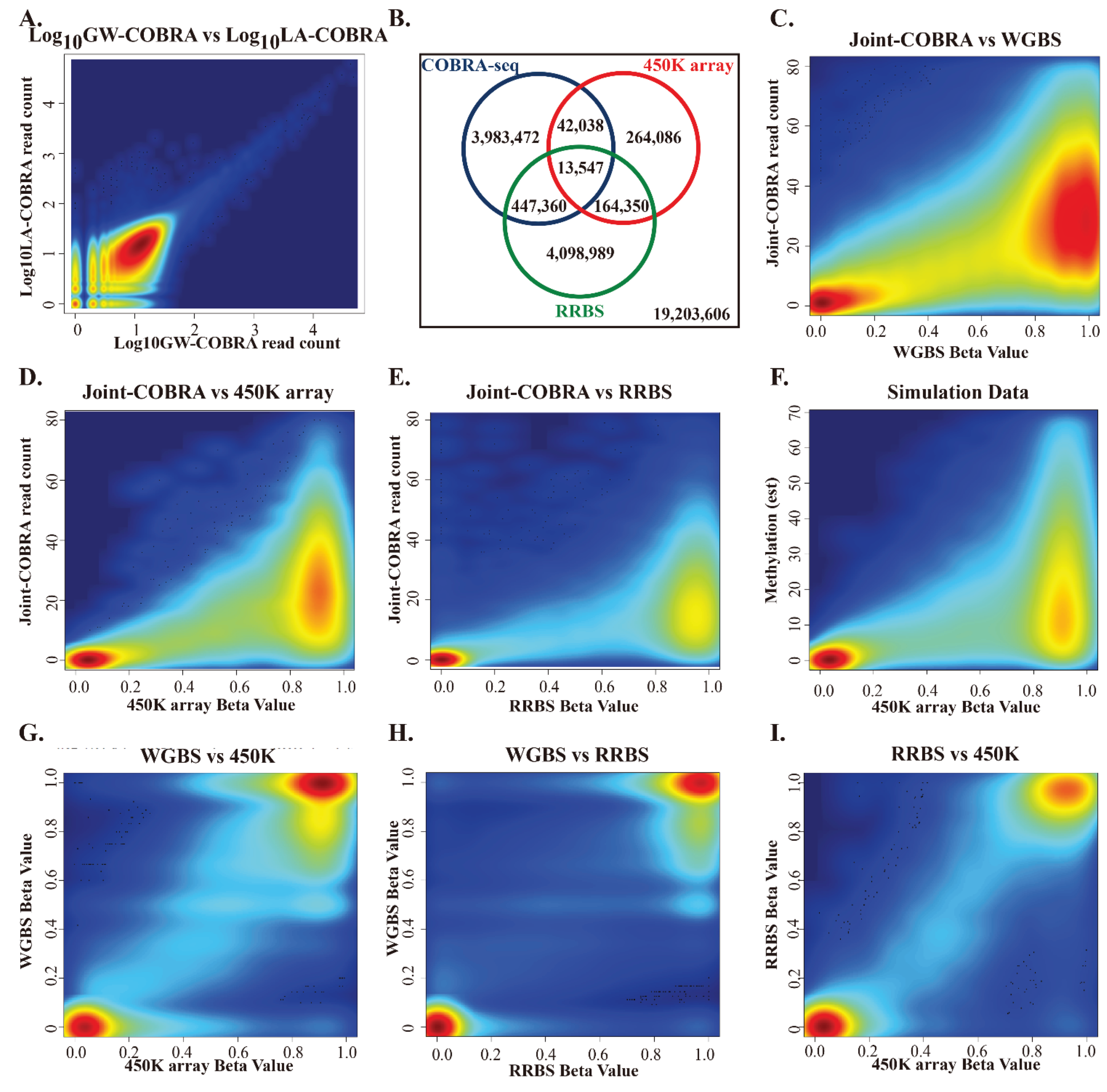
3.2. Number of GW-COBRA and LA-COBRA Reads and Mapping to Genome

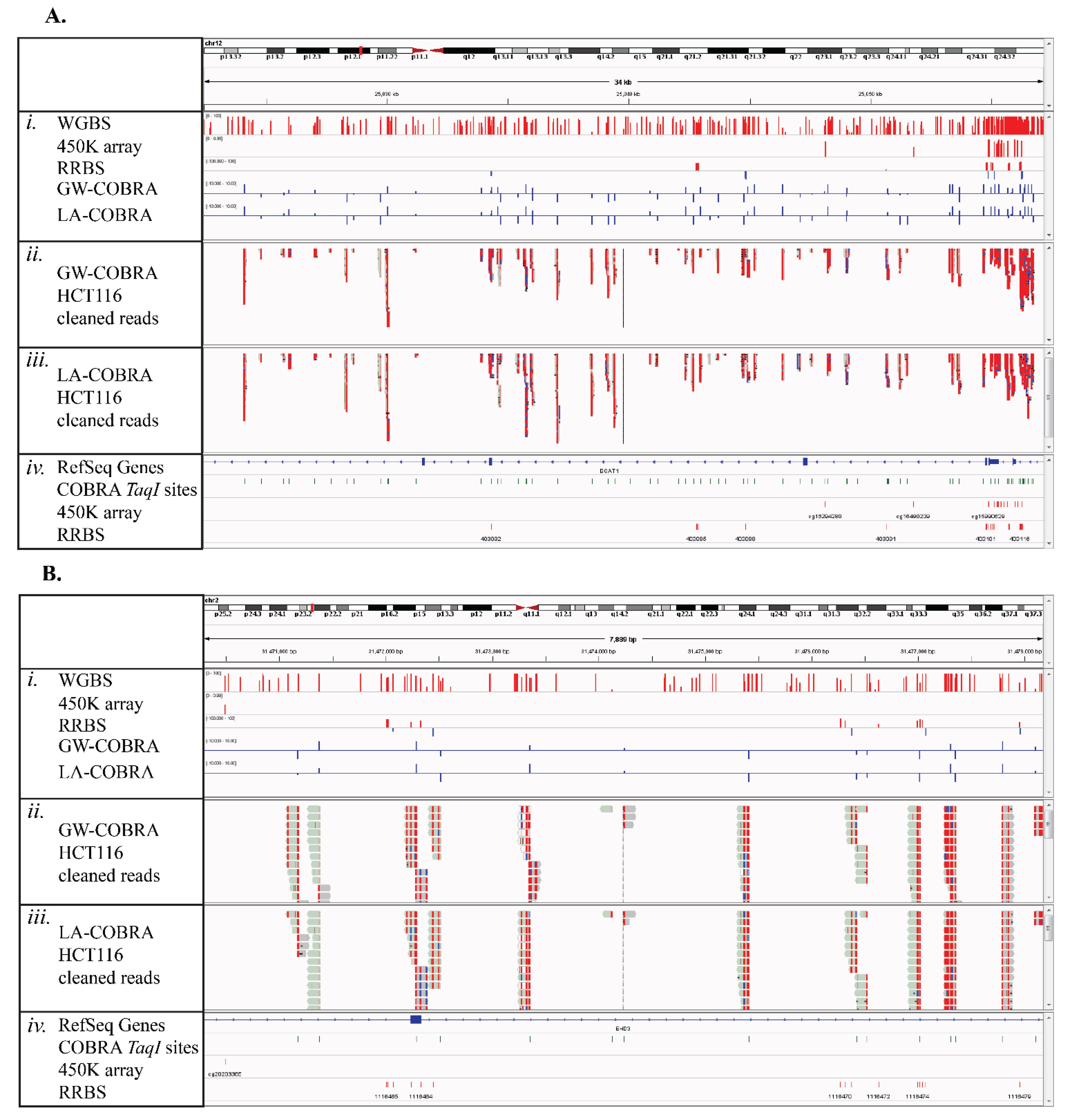
3.3. Comparison of HCT116 COBRA-Seq with RRBS and 450K Array Data
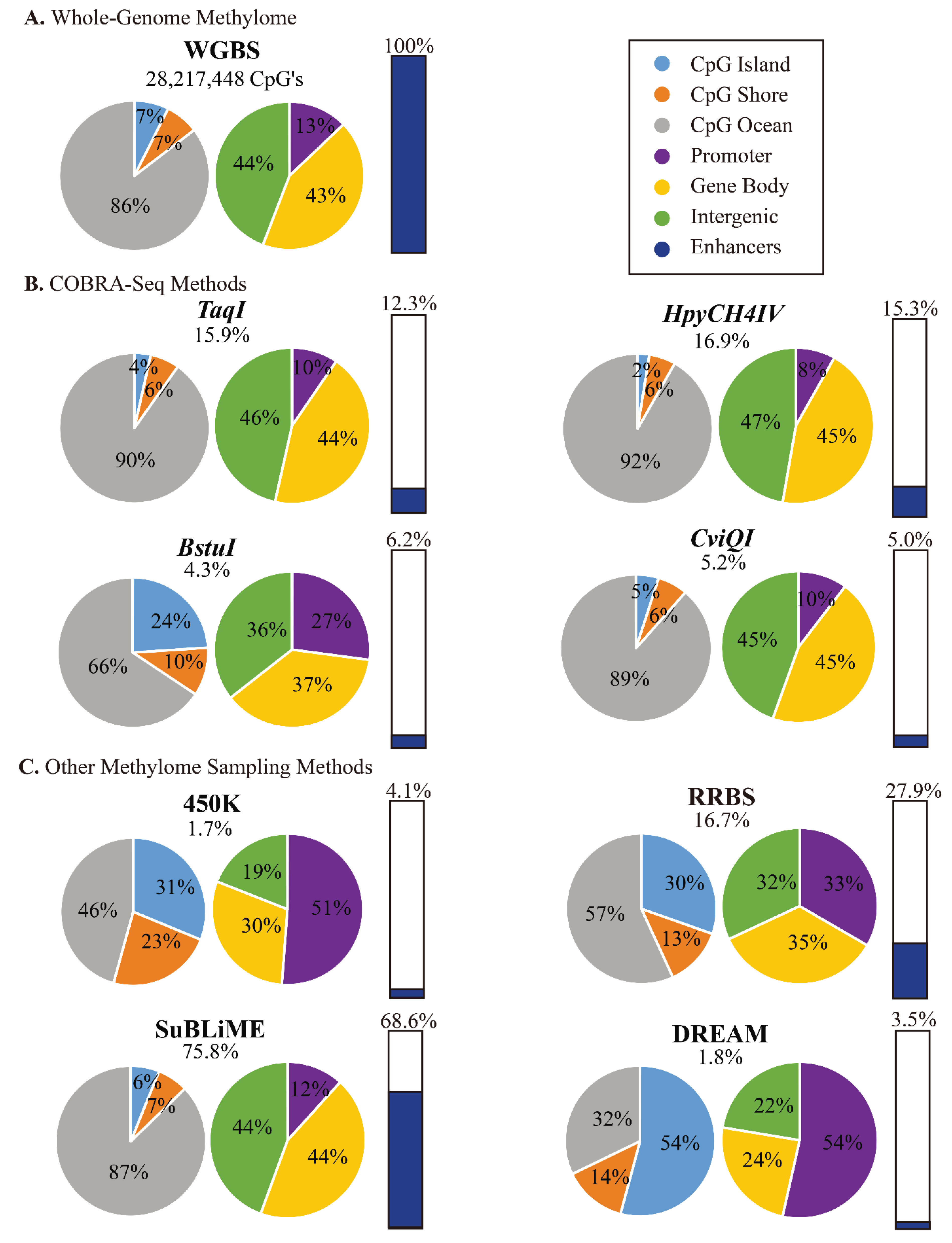
3.4. COBRA-Seq Features

3.5. Comparison of COBRA-Seq Features with Other Methods of Methylome Sampling

| Methylome Methods | Complexity Reduction Type | M or M + U Fraction | Methylome Sampling (Yes/No) | Favour of Enrichment Towards | Comments |
|---|---|---|---|---|---|
| WGBS [1] | N/A | M + U | N/A | N/A | High cost, can detect non-CpG methylation, genomic input* (0.05–0.1 μg). |
| MBDCap-Seq [34,62], MIRA-Seq [38] | Affinity capture | M | No | CpG-rich | Dependent on CpG density, effected by salt concentration, covers about 18% of the CpGs [24], 28,500 CpG islands [34], DNA input = 0.2–1 μg. |
| MeDIP [63], MeDIP-Seq [36] | Affinity capture | M | No | CpG-rich | Bias towards 5mC-rich regions, Captures single-stranded DNA, prone to technical variability, coverage is read-depth dependent, input = 0.15–5 μg. |
| SuBLiME [42] | Methylated cytosine capture | M | Yes | CpG-rich and poor | Substitutes biotin-14-dCTP or biotin-14-dGTP at the position of the 5mC in bisulfite treated DNA, input = 2 μg. |
| COBRA-seq | Restriction digest | M | Yes | CpG-rich and poor | Provides relative methylation levels like MeDIP/MBD-Cap, 1%–17% of the CpGs for single digest, can detect non-CpG methylation, input = 0.1–1 μg. |
| Nimblegen SeqCap [39] | Hybridization capture | M + U | Yes | CpG-rich and poor | “Off-the-shelf” version for human genome only/similar regions covered as 450K array, can be customized [39], can detect non-CpG methylation, input = 0.5–1 μg. |
| Agilent SureSelect [64] | Hybridization capture | M + U | Yes | CpG-rich and poor | Available for human genome only, covers 3.7 million CpG sites, input = 0.5 μg. |
| 450K array [30,65] | Microarray | M + U | Yes | CpG-rich | Arrays comes in 12 per slide, available only for humans, not readily customized, input = 0.5–1 μg. |
| RRBS [35,53] | Restriction digest | M + U | Yes | CpG-rich/medium | Can detect non-CpG methylation, input = 0.1–0.3 μg. |
| Methyl-Seq [33], HELP-Seq [66] | Restriction digest | M + U | Yes | CpG-rich | Assesses 0.25 to 1.3 million CCGG sites in human genome by difference in read fractions in HpaII vs. MspI libraries, input = 0.01–0.1 μg. |
| CHARM [32,67] | Restriction digest | M + U | Yes | CpG-rich and poor | Array-based and available for human, mice and rat, assesses 3.5 to 7 million CpG sites, input = 5 μg. |
| DREAM [37] | Restriction digest | M + U | Yes | CpG-rich | Assesses methylation at ~0.15 million sites in human genome by sequential SmaI/XmaI digestion and library sequencing, input = 5 μg. |
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Novik, K.L.; Nimmrich, I.; Genc, B.; Maier, S.; Piepenbrock, C.; Olek, A.; Beck, S. Epigenomics: Genome-wide study of methylation phenomena. Curr. Issues Mol. Biol. 2002, 4, 111–128. [Google Scholar] [PubMed]
- Rakyan, V.K.; Hildmann, T.; Novik, K.L.; Lewin, J.; Tost, J.; Cox, A.V.; Andrews, T.D.; Howe, K.L.; Otto, T.; Olek, A.; et al. DNA methylation profiling of the human major histocompatibility complex: A pilot study for the human epigenome project. PLoS Biol. 2004, 2, e405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun bisulphite sequencing of the arabidopsis genome reveals DNA methylation patterning. Nature 2008, 452, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Cokus, S.J.; Zhang, X.; Chen, P.Y.; Bostick, M.; Goll, M.G.; Hetzel, J.; Jain, J.; Strauss, S.H.; Halpern, M.E.; et al. Conservation and divergence of methylation patterning in plants and animals. Proc. Natl. Acad. Sci. USA 2010, 107, 8689–8694. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; O’Malley, R.C.; Tonti-Filippini, J.; Gregory, B.D.; Berry, C.C.; Millar, A.H.; Ecker, J.R. Highly integrated single-base resolution maps of the epigenome in arabidopsis. Cell 2008, 133, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Stroud, H.; Greenberg, M.V.; Feng, S.; Bernatavichute, Y.V.; Jacobsen, S.E. Comprehensive analysis of silencing mutants reveals complex regulation of the arabidopsis methylome. Cell 2013, 152, 352–364. [Google Scholar] [CrossRef] [PubMed]
- Zemach, A.; McDaniel, I.E.; Silva, P.; Zilberman, D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 2010, 328, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Zilberman, D.; Gehring, M.; Tran, R.K.; Ballinger, T.; Henikoff, S. Genome-wide analysis of arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat. Genet. 2007, 39, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhu, J.; Hu, F.; Ge, S.; Ye, M.; Xiang, H.; Zhang, G.; Zheng, X.; Zhang, H.; Zhang, S.; et al. Single-base resolution maps of cultivated and wild rice methylomes and regulatory roles of DNA methylation in plant gene expression. BMC Genomics 2012. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.; Choi, J.; Lee, G.W.; Park, S.Y.; Huh, A.; Dean, R.A.; Lee, Y.H. Genome-wide profiling of DNA methylation provides insights into epigenetic regulation of fungal development in a plant pathogenic fungus, magnaporthe oryzae. Sci. Rep. 2015. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.Y.; Lin, J.Q.; Wu, H.L.; Wang, C.C.; Huang, S.J.; Luo, Y.F.; Sun, J.H.; Zhou, J.X.; Yan, S.J.; He, J.G.; et al. Bisulfite sequencing reveals that aspergillus flavus holds a hollow in DNA methylation. PLoS ONE 2012, 7, e30349. [Google Scholar] [CrossRef] [PubMed]
- Selker, E.U.; Tountas, N.A.; Cross, S.H.; Margolin, B.S.; Murphy, J.G.; Bird, A.P.; Freitag, M. The methylated component of the neurospora crassa genome. Nature 2003, 422, 893–897. [Google Scholar] [CrossRef] [PubMed]
- Salzberg, A.; Fisher, O.; Siman-Tov, R.; Ankri, S. Identification of methylated sequences in genomic DNA of adult drosophila melanogaster. Biochem. Biophys. Res. Commun. 2004, 322, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Takayama, S.; Dhahbi, J.; Roberts, A.; Mao, G.; Heo, S.J.; Pachter, L.; Martin, D.I.; Boffelli, D. Genome methylation in D. melanogaster is found at specific short motifs and is independent of DNMT2 activity. Genome Res. 2014, 24, 821–830. [Google Scholar] [CrossRef] [PubMed]
- Lyko, F.; Foret, S.; Kucharski, R.; Wolf, S.; Falckenhayn, C.; Maleszka, R. The honey bee epigenomes: Differential methylation of brain DNA in queens and workers. PLoS Biol. 2010, 8, e1000506. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Zhang, J.; Wang, J.J.; Wang, L.; Zhang, L.; Li, G.; Yang, X.; Ma, X.; Sun, X.; Cai, J.; et al. Sperm, but not oocyte, DNA methylome is inherited by zebrafish early embryos. Cell 2013, 153, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Potok, M.E.; Nix, D.A.; Parnell, T.J.; Cairns, B.R. Reprogramming the maternal zebrafish genome after fertilization to match the paternal methylation pattern. Cell 2013, 153, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Habibi, E.; Brinkman, A.B.; Arand, J.; Kroeze, L.I.; Kerstens, H.H.; Matarese, F.; Lepikhov, K.; Gut, M.; Brun-Heath, I.; Hubner, N.C.; et al. Whole-genome bisulfite sequencing of two distinct interconvertible DNA methylomes of mouse embryonic stem cells. Cell Stem Cell 2013, 13, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Meissner, A.; Mikkelsen, T.S.; Gu, H.; Wernig, M.; Hanna, J.; Sivachenko, A.; Zhang, X.; Bernstein, B.E.; Nusbaum, C.; Jaffe, D.B.; et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 2008, 454, 766–770. [Google Scholar] [CrossRef] [PubMed]
- Doherty, R.; Couldrey, C. Exploring genome wide bisulfite sequencing for DNA methylation analysis in livestock: A technical assessment. Front. Genet. 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloyd, V.K.; Brisson, J.A.; Fitzpatrick, K.A.; McEachern, L.A.; Verhulst, E.C. The epigenetics of emerging and nonmodel organisms. Genet. Res. Int. 2012. [Google Scholar] [CrossRef] [PubMed]
- Perfus-Barbeoch, L.; Castagnone-Sereno, P.; Reichelt, M.; Fneich, S.; Roquis, D.; Pratx, L.; Cosseau, C.; Grunau, C.; Abad, P. Elucidating the molecular bases of epigenetic inheritance in non-model invertebrates: The case of the root-knot nematode meloidogyne incognita. Front. Physiol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Stirzaker, C.; Taberlay, P.C.; Statham, A.L.; Clark, S.J. Mining cancer methylomes: Prospects and challenges. Trends Genet. 2014, 30, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Frommer, M.; Mcdonald, L.E.; Millar, D.S.; Collis, C.M.; Watt, F.; Grigg, G.W.; Molloy, P.L.; Paul, C.L. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. USA 1992, 89, 1827–1831. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.J.; Harrison, J.; Paul, C.L.; Frommer, M. High sensitivity mapping of methylated cytosines. Nucleic Acids Res. 1994, 22, 2990–2997. [Google Scholar] [PubMed]
- Ziller, M.J.; Gu, H.C.; Muller, F.; Donaghey, J.; Tsai, L.T.Y.; Kohlbacher, O.; de Jager, P.L.; Rosen, E.D.; Bennett, D.A.; Bernstein, B.E.; et al. Charting a dynamic DNA methylation landscape of the human genome. Nature 2013, 500, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef] [PubMed]
- Glastad, K.M.; Hunt, B.G.; Goodisman, M.A.D. Evolutionary insights into DNA methylation in insects. Curr. Opin. Insect Sci. 2014, 1, 25–30. [Google Scholar] [CrossRef]
- Sandoval, J.; Heyn, H.A.; Moran, S.; Serra-Musach, J.; Pujana, M.A.; Bibikova, M.; Esteller, M. Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics 2011, 6, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Bibikova, M.; Lin, Z.; Zhou, L.; Chudin, E.; Garcia, E.W.; Wu, B.; Doucet, D.; Thomas, N.J.; Wang, Y.; Vollmer, E.; et al. High-throughput DNA methylation profiling using universal bead arrays. Genome Res. 2006, 16, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, R.A.; Ladd-Acosta, C.; Carvalho, B.; Wu, H.; Brandenburg, S.A.; Jeddeloh, J.A.; Wen, B.; Feinberg, A.P. Comprehensive high-throughput arrays for relative methylation (CHARM). Genome Res. 2008, 18, 780–790. [Google Scholar] [CrossRef] [PubMed]
- Brunner, A.L.; Johnson, D.S.; Kim, S.W.; Valouev, A.; Reddy, T.E.; Neff, N.F.; Anton, E.; Medina, C.; Nguyen, L.; Chiao, E.; et al. Distinct DNA methylation patterns characterize differentiated human embryonic stem cells and developing human fetal liver. Genome Res. 2009, 19, 1044–1056. [Google Scholar] [CrossRef] [PubMed]
- Serre, D.; Lee, B.H.; Ting, A.H. Mbd-isolated genome sequencing provides a high-throughput and comprehensive survey of DNA methylation in the human genome. Nucleic Acids Res. 2010, 38, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Ziller, M.J.; Muller, F.; Liao, J.; Zhang, Y.Y.; Gu, H.C.; Bock, C.; Boyle, P.; Epstein, C.B.; Bernstein, B.E.; Lengauer, T.; et al. Genomic distribution and inter-sample variation of non-CpG methylation across human cell types. PLoS Genet. 2011, 7, e1002389. [Google Scholar] [CrossRef] [PubMed]
- Taiwo, O.; Wilson, G.A.; Morris, T.; Seisenberger, S.; Reik, W.; Pearce, D.; Beck, S.; Butcher, L.M. Methylome analysis using MeDIP-seq with low DNA concentrations. Nat. Protoc. 2012, 7, 617–636. [Google Scholar] [CrossRef] [PubMed]
- Jelinek, J.; Liang, S.D.; Lu, Y.; He, R.; Ramagli, L.S.; Shpall, E.J.; Estecio, M.R.H.; Issa, J.P.J. Conserved DNA methylation patterns in healthy blood cells and extensive changes in leukemia measured by a new quantitative technique. Epigenetics 2012, 7, 1368–1378. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Kadam, S.; Xiong, W.; Rauch, T.A.; Jin, S.G.; Pfeifer, G.P. MIRA-seq for DNA methylation analysis of CpG islands. Epigenomics 2015. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Suzuki, M.; Wendt, J.; Patterson, N.; Eichten, S.R.; Hermanson, P.J.; Green, D.; Jeddeloh, J.; Richmond, T.; Rosenbaum, H.; et al. Post-conversion targeted capture of modified cytosines in mammalian and plant genomes. Nucleic Acids Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Laird, P.W. Principles and challenges of genomewide DNA methylation analysis. Nat. Rev. Genet. 2010, 11, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Z.; Laird, P.W. COBRA: A sensitive and quantitative DNA methylation assay. Nucleic Acids Res. 1997, 25, 2532–2534. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.P.; Shaw, J.M.; Molloy, P.L. Identification of differentially methylated regions using streptavidin bisulfite ligand methylation enrichment (SuBLiME), a new method to enrich for methylated DNA prior to deep bisulfite genomic sequencing. Epigenetics 2013, 8, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Rybak, J.N.; Scheurer, S.B.; Neri, D.; Elia, G. Purification of biotinylated proteins on streptavidin resin: A protocol for quantitative elution. Proteomics 2004, 4, 2296–2299. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, W.A.; Bartfai, R.; Francoijs, K.J.; Stunnenberg, H.G. Linear amplification for deep sequencing. Nat. Protoc. 2011, 6, 1026–1036. [Google Scholar] [CrossRef] [PubMed]
- CSIRO Data Access Portal. Available online: https://data.Csiro.Au/dap/home?Execution=e1s1 (accessed on 30 september 2015).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011. [Google Scholar] [CrossRef]
- Pedersen, B.S.; Eyring, K.; De, S.; Yang, I.V.; Schwartz, D.A. Fast and Accurate Alignment of Long Bisulfite-Seq Reads. Available online: http://arxiv.org/ (accessed on 30 September 2015).
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with Bwa-Mem. Available online: http://arxiv.org/ (accessed on 30 September 2015).
- Lizio, M.; Harshbarger, J.; Shimoji, H.; Severin, J.; Kasukawa, T.; Sahin, S.; Abugessaisa, I.; Fukuda, S.; Hori, F.; Ishikawa-Kato, S.; et al. Gateways to the FANTOM5 promoter level mammalian expression atlas. Genome Biol. 2015. [Google Scholar] [CrossRef] [PubMed]
- COBRA-Seq: Sensitive and Quantitative Methylome Profiling. Available online: https://stash.Csiro.Au/users/ros259/repos/cobra-seq (accessed on 30 September 2015).
- Ji, L.; Sasaki, T.; Sun, X.; Ma, P.; Lewis, Z.A.; Schmitz, R.J. Methylated DNA is over-represented in whole-genome bisulfite sequencing data. Front. Genet. 2014. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Brieba, L.G.; Sousa, R. Misincorporation by wild-type and mutant t7 RNA polymerases: Identification of interactions that reduce misincorporation rates by stabilizing the catalytically incompetent open conformation. Biochemistry 2000, 39, 11571–11580. [Google Scholar] [CrossRef] [PubMed]
- Smith, Z.D.; Gu, H.C.; Bock, C.; Gnirke, A.; Meissner, A. High-throughput bisulfite sequencing in mammalian genomes. Methods 2009, 48, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Blattler, A.; Yao, L.; Witt, H.; Guo, Y.; Nicolet, C.M.; Berman, B.P.; Farnham, P.J. Global loss of DNA methylation uncovers intronic enhancers in genes showing expression changes. Genome Biol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Akalin, A.; Garrett-Bakelman, F.E.; Kormaksson, M.; Busuttil, J.; Zhang, L.; Khrebtukova, I.; Milne, T.A.; Huang, Y.S.; Biswas, D.; Hess, J.L.; et al. Base-pair resolution DNA methylation sequencing reveals profoundly divergent epigenetic landscapes in acute myeloid leukemia. PLoS Genet. 2012, 8, e1002781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, S.K.; Baker, R.T.; McEvoy, A.; Murray, D.H.; Thomas, M.; Molloy, P.L.; Mitchell, S.; Lockett, T.; Young, G.P.; LaPointe, L.C. A two-gene blood test for methylated DNA sensitive for colorectal cancer. PLoS ONE 2015, 10, e0125041. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.M.; Ross, J.P.; Drew, H.R.; Ho, T.; Brown, G.S.; Saunders, N.F.; Duesing, K.R.; Buckley, M.J.; Dunne, R.; Beetson, I.; et al. A panel of genes methylated with high frequency in colorectal cancer. BMC Cancer 2014. [Google Scholar] [CrossRef] [PubMed]
- Davey, J.W.; Blaxter, M.L. RADSeq: Next-generation population genetics. Brief. Funct. Genomics 2010, 9, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Ruike, Y.; Imanaka, Y.; Sato, F.; Shimizu, K.; Tsujimoto, G. Genome-wide analysis of aberrant methylation in human breast cancer cells using methyl-DNA immunoprecipitation combined with high-throughput sequencing. BMC Genomics 2010. [Google Scholar] [CrossRef] [PubMed]
- Ulahannan, N.; Greally, J.M. Genome-wide assays that identify and quantify modified cytosines in human disease studies. Epigenetics Chromatin 2015. [Google Scholar] [CrossRef] [PubMed]
- Adusumalli, S.; Mohd Omar, M.F.; Soong, R.; Benoukraf, T. Methodological aspects of whole-genome bisulfite sequencing analysis. Brief. Bioinform. 2015, 16, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; Stirzaker, C.; Statham, A.L.; Coolen, M.W.; Song, J.Z.; Nair, S.S.; Strbenac, D.; Speed, T.P.; Clark, S.J. Evaluation of affinity-based genome-wide DNA methylation data: Effects of CpG density, amplification bias, and copy number variation. Genome Res. 2010, 20, 1719–1729. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Davies, J.J.; Wittig, D.; Oakeley, E.J.; Haase, M.; Lam, W.L.; Schubeler, D. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat. Genet. 2005, 37, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Gnirke, A.; Melnikov, A.; Maguire, J.; Rogov, P.; LeProust, E.M.; Brockman, W.; Fennell, T.; Giannoukos, G.; Fisher, S.; Russ, C.; et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat. Biotechnol. 2009, 27, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Dedeurwaerder, S.; Defrance, M.; Calonne, E.; Denis, H.; Sotiriou, C.; Fuks, F. Evaluation of the infinium methylation 450K technology. Epigenomics 2011, 3, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Oda, M.; Glass, J.L.; Thompson, R.F.; Mo, Y.; Olivier, E.N.; Figueroa, M.E.; Selzer, R.R.; Richmond, T.A.; Zhang, X.; Dannenberg, L.; et al. High-resolution genome-wide cytosine methylation profiling with simultaneous copy number analysis and optimization for limited cell numbers. Nucleic Acids Res. 2009, 37, 3829–3839. [Google Scholar] [CrossRef] [PubMed]
- Ordway, J.M.; Bedell, J.A.; Citek, R.W.; Nunberg, A.; Garrido, A.; Kendall, R.; Stevens, J.R.; Cao, D.; Doerge, R.W.; Korshunova, Y.; et al. Comprehensive DNA methylation profiling in a human cancer genome identifies novel epigenetic targets. Carcinogenesis 2006, 27, 2409–2423. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Arguelles, D.B.; Lee, S.; Papadopoulos, V. In silico analysis identifies novel restriction enzyme combinations that expand reduced representation bisulfite sequencing CpG coverage. BMC Res. Notes 2014. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xia, Y.; Li, L.; Gong, D.; Yao, Y.; Luo, H.; Lu, H.; Yi, N.; Wu, H.; Zhang, X.; et al. Double restriction-enzyme digestion improves the coverage and accuracy of genome-wide CpG methylation profiling by reduced representation bisulfite sequencing. BMC Genomics 2013. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varinli, H.; Statham, A.L.; Clark, S.J.; Molloy, P.L.; Ross, J.P. COBRA-Seq: Sensitive and Quantitative Methylome Profiling. Genes 2015, 6, 1140-1163. https://doi.org/10.3390/genes6041140
Varinli H, Statham AL, Clark SJ, Molloy PL, Ross JP. COBRA-Seq: Sensitive and Quantitative Methylome Profiling. Genes. 2015; 6(4):1140-1163. https://doi.org/10.3390/genes6041140
Chicago/Turabian StyleVarinli, Hilal, Aaron L. Statham, Susan J. Clark, Peter L. Molloy, and Jason P. Ross. 2015. "COBRA-Seq: Sensitive and Quantitative Methylome Profiling" Genes 6, no. 4: 1140-1163. https://doi.org/10.3390/genes6041140