1. Introduction
Eugenol (4-allyl-2-methoxyphenol), a major component of clove oil, is widely used as a fragrance and flavoring agent in cosmetic, food and pharmacological products [
1,
2]. In addition, studies have shown that the therapeutic use of eugenol has great potential as antibiotic, anticarcinogenic, antimicrobial, antioxidant, anti-inflammatory, fungicidal, larvicidal and other agents [
3,
4,
5,
6,
7,
8]. However, in high concentrations, it has an adverse effect of provoking inflammatory and allergic reactions, possibly due to its low instability, which favors the formation of phenoxyl radicals via its pro-oxidant activity [
2,
9]. Despite the fact that its structure is polyfunctional and readily accessible, eugenol is considered as a useful starting material for producing valuable derivatives through chemical synthesis [
2,
10,
11,
12,
13,
14,
15] or biochemistry [
1,
4,
9,
16,
17,
18].
Eugenol derivatives can be obtained by reactions on the hydroxyl group of its structure via acetylation [
4,
15,
19] or benzoylation [
2,
9,
20]. Investigations have shown that this modification also improves antioxidant and larvicidal activities 2 or 5 times, respectively, moreover to the reduced level of cytotoxicity compared to its precursor, eugenol [
4,
7]. Futhermore, studies have found that small amounts of eugenyl acetate are effective against the development of larvae of the
Aedes aegypti mosquito [
4,
14,
21]. The results reported in the literature ensure the potential application of eugenyl acetate in formulations of insecticides [
4,
7].
The
Aedes aegypti mosquito is the virus vector of diseases such as dengue fever, yellow fever, Chikungunya, zika and cases of microcephaly in newborns [
4,
22]. For dengue disease, there are no drugs or vaccines available to prevent the spread of the virus so that the remaining method of controlling the propagation of its vector through the use of insecticides to prevent adult mosquito infestation or the use of larvicides synthetics such as organophosphates, organochlorines and others [
6,
22]. Mosquito population control is more effective in the larval stage than in adulthood [
21]. The eugenyl acetate, besides being an efficient larvicide, still complies with the principles of green chemistry because it comes from biomass [
4,
6,
7,
14,
21].
Most of the time the current chemical route to synthesize eugenol esters is quite problematic by making use of homogeneous catalysis [
13,
21,
23,
24], causing serious problems of reactor corrosion, volatile generation of unwanted effluents, impurity and unsatisfactory yield of the product, as well as a possible adverse impact on the environment, and in this sense the search for alternative heterogeneous catalysts has been constant [
2,
9,
20]. Biocatalytic processes to obtain the ester, in spite of being alternative and environmentally friendly, in many cases still present high costs, mainly when immobilized, besides the low reuse capacity [
1,
10,
16].
Thus, the use of heterogeneous catalysts is particularly advantageous because they are versatile, possess high operational efficiency under moderate reaction conditions, and additionally they provide a more ecological and economical route for esterification reactions [
25,
26,
27,
28,
29,
30,
31]. In recent years, some acidic solids have been used as catalysts for the esterification or acetylation of eugenol, for example modified acid zirconia (UDCaT-5) [
2] and ionic resins [
10,
11,
12], which not only can be easily recovered and reused, but also favored the development of continuous production processes for a series of industrially important productions.
Heteropolyacids (HPAs), especially those of the Keggin series, are widely used as catalysts for the synthesis of fine and specialized chemicals [
32,
33,
34,
35,
36]. Due to their strong acidity, they generally exhibit higher catalytic activities than conventional catalysts, such as mineral acids, ion exchange resins, mixed oxides, zeolites, etc. Also, HPA catalysis has no side reactions, such as sulfonation and chlorination, which often occur with mineral acids [
34].
In the literature there are reports that the solid HPA, used as a homogeneous catalyst, and silica-supported HPAs acting as heterogeneous catalysts, are efficient for isomerization reactions of α-pinene oxides [
34], limonene [
32], cyclization of nerolidol and farnesol [
36] and aldehyde cycloaddition [
35]. However, studies on the use of heteropoly acids supported in mesoporous materials for the production of eugenyl acetate are not reported.
The nature of the support is an important factor for a successful reaction towards the best catalytic activity [
37]. MCM-41 is the most studied member of the M41S family applied as catalytic support because of its regular structure, large surface area and uniform pore size, ranging from 15 to 100 Å [
30,
38,
39,
40,
41]. The structure of the MCM-41 represents a hexagonal arrangement of cylindrical channels consisting of amorphous silica [
41]. In the synthesis of MCM-41 the structure-directing agent cetyltrimethylammonium bromide (CTABr) and the tetraethylorthosilicate (TEOS) are the source of silica generally used. TEOS besides being toxic has a high cost [
42,
43]. Due to economic and environmental issues, research has been intensified by new sources of low-cost silica, such as metakaolin, for the synthesis of ordered mesoporous materials [
44,
45,
46].
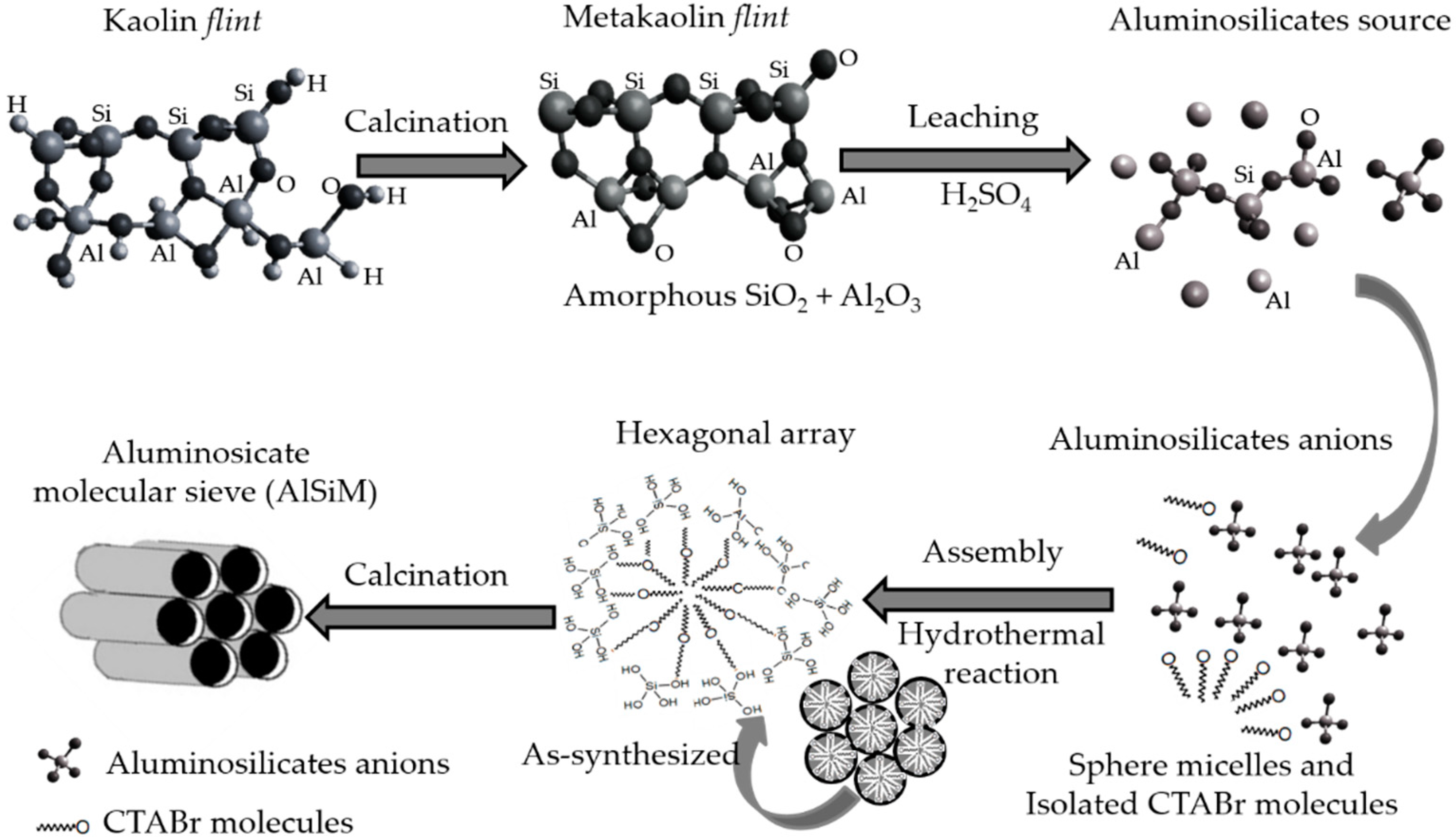
The Amazon
flint kaolin (KF) is an industrial waste disposed of in the mine after the exploitation of soft kaolin, generating a considerable environmental impact, being constituted mainly by kaolinite clay, presenting high levels of silicon, aluminum and iron [
25,
47,
48]. Our research group has aimed to add value to wasted kaolinite as alternative to minimize environmental impacts. Previous studies showed wasted kaolinite as feedstock for catalyst synthesis [
25,
26,
29,
31], catalytic supports [
30,
49] and for mesoporous aluminosilicate synthesis [
27], for all cases, they were efficient for the esterification of free fatty acids, directed to the field of the production of biofuels [
25,
26,
27,
28,
29,
30,
31]. The
flint kaolin also shows potential as a starting material for zeolite synthesis [
47].
In this sense, the objective of this study was to investigate an alternative to minimize problems caused by
flint kaolin in mining, using this waste as a source of silica and aluminum in the synthesis of AlSiM. This work is the first report on the valorization of
flint kaolin containing Al, Si, and a high content of Fe and Ti and other metals in small quantities for which pretreatment and purification steps [
27] before the AlSiM synthesis.
In this work, also for the first time, the effect of AlSiM support on the catalytic activity of HPMo for the acetylation of eugenol was investigated. The support and catalysts synthesized were characterized by several physico-chemical techniques. The catalytic activity was evaluated for the production of eugenyl acetate by acetylation of eugenol. The influence of several reaction parameters (such as catalyst concentration, eugenol molar ratio/acetic anhydride, temperature and reaction time) in the catalytic performance was also studied. A kinetic study was performed and the order of reaction, reaction rate constant and apparent activation energy were determined as well as a catalyst recycling study was realized. As far as we know, no attempt to use HPA catalysts anchored in mesoporous silicates for this reaction has been made before.
2. Materials and Methods
2.1. Raw Material and Chemicals
12-molybdophosphoric acid (H3PMo12O40.H2O, HPMo, Mo = 63.08%, VETEC, Darmstadt, Germany), sulfuric acid (H2SO4, 98%, ISOFAR, Duque de Caxias, Rio de Janeiro, Brazil) and hydrochloric acid (HCl, 37%, Fmaia, Belo Horizonte, MG, Brazil), cetyltrimethylammonium bromide (C16H33(CH3), CTABr, Vetec, Rio de Janeiro, Brazil), ethanol (EtOH, 98%, Nuclear, São Paulo, SP–Brazil) and sodium hydroxide (NaOH, VETEC, Rio de Janeiro, Brazil), eugenol (EugOH, 99% Sigma Aldrich, Darmstadt, Germany) and acetic anhydride (AA, Nuclear, São Paulo, SP–Brazil). All chemical reagents and solvents were analytical grade and used without further purification. The flint kaolin was kindly provided by the UFPA Institute of Geology from the Capim River Region (Pará Brazil).
2.2. Preparation of Catalysts
2.2.1. Thermal and Chemical Treatment of Amazon Flint Kaolin
The
flint kaolin was triturated, and the sandy fraction was separated for retention in a sieve. The fraction below 62 μm particle size was then collected, diluted in distilled water and centrifuged for separation of the silt fraction, thereby obtaining the clay fraction [
25]. The
flint kaolin was calcined at 750 °C for 5 h to give the metakaolinite (MF) phase which is amorphous. This material was then leached at 90 °C for 1 h with 2.5 mols L
−1 H
2SO
4 solution in a MF/H
2SO
4 molar ratio of 1:10. After leaching, the product was filtered, washed with 100 mL of 0.5 mols L
−1 H
2SO
4 solution and with water to neutral pH, and then dried at 120 °C for 12 h. The sample obtained after the acid leaching was denominated MFL (
Scheme 1). All of these procedures are as described by Lima et al. [
27].
2.2.2. Synthesis of Mesoporous Aluminosilicate (AlSiM) by the Hydrothermal Method
The AlSiM synthesis procedure was based on literature methodology [
27] where 3 g of the MFL was added in a solution composed of 1.8168 g of CTABr, 0.6172 g of NaOH and 134.7 mL of distilled water which was stirred for 24 h. After this period the solution was hydrothermally treated in a stainless steel autoclave-coated teflon vessel at 110 °C for 24 h. The formed hydrogel was filtered and washed with distilled water and dried at 110 °C for 12 h and calcined at 540 °C for 5 h for removal of the surfactant (CTABr). Based on the procedures described above, it was proposed a possible process for the preparation of ordered mesoporous AlSiM material from natural kaolin
flint and is summarized in according to
Scheme 1 adapted from literature [
42,
43,
50].
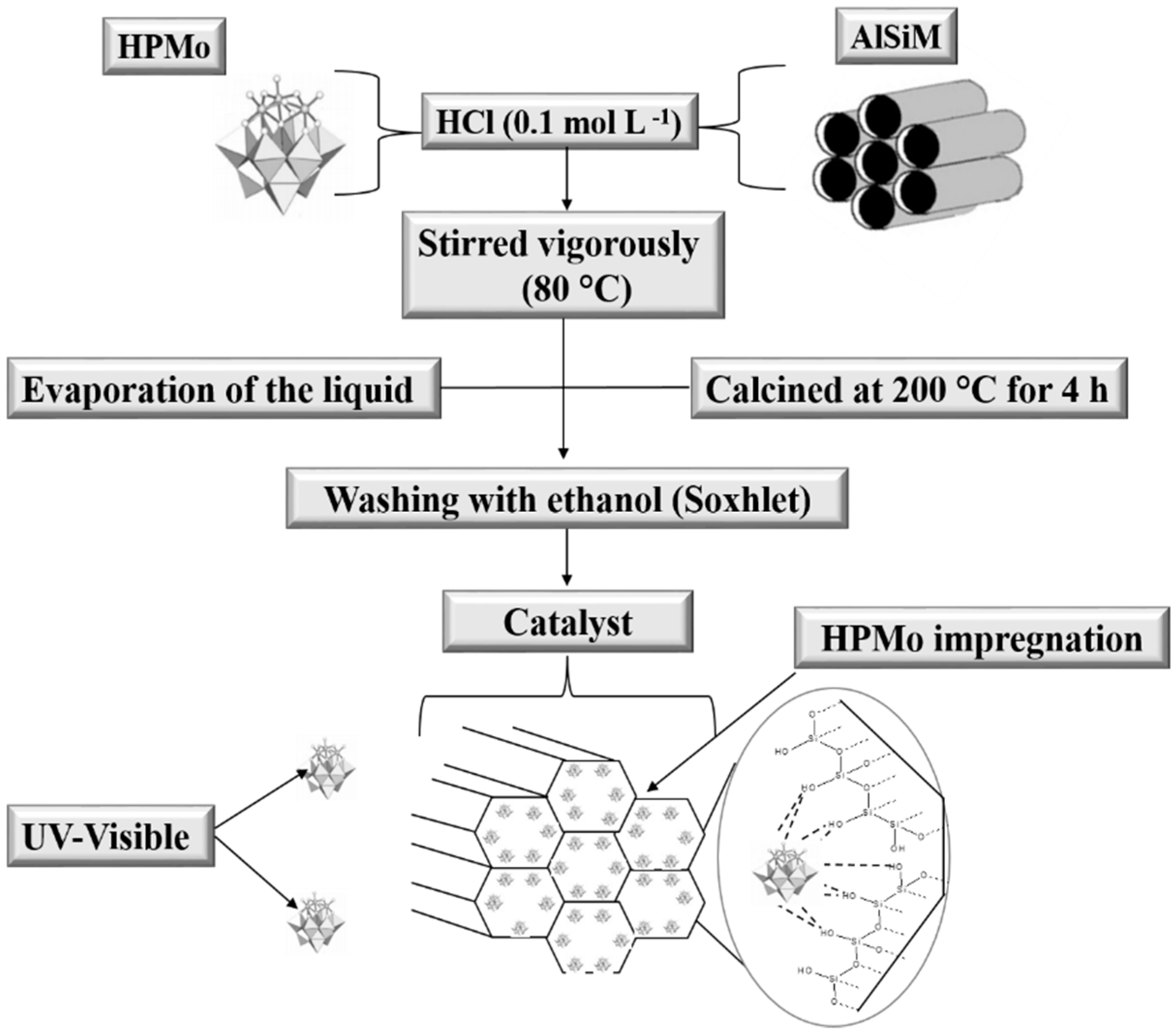
2.2.3. Catalyst Synthesis (HPMo Anchored to AlSiM)
The HPMo anchored to the AlSiM was synthesized by impregnation according to Pires et al. [
30] and Oliveira et al. [
28]. Each gram of AlSiM was dispersed under vigorous stirring in a 0.1 mol L
−1 aqueous HCl solution, which already contained the desired amounts of HPMo (0.11 g, 0.25 g and 0.43 g). The resulting mixture of HPMo and AlSiM was heated at 80 °C under constant stirring until complete evaporation of water. The solid obtained was pulverized and dried at 120 °C for 12 h and finally calcined at 200 °C for 4 h at a heating rate of 10 °C min
−1. The samples obtained were designated as xHPMo/AlSiM, where x refers to the percentage of HPMo supported in the AlSiM, which varied in 10%, 20% and 30% by mass, according to
Scheme 2 [
28].
2.2.4. Washing of AlSiM Anchored with HPMo
To eliminate excess HPMo on the surface of the material, which could contribute to homogeneous catalysis during the reaction, the xHPMo/AlSiM was subjected to a washing step. The xHPMo/AlSiM (3 g) was inserted into a Soxhlet extractor where it was washed with ethanol (300 mL) under constant reflux for 24 h, according to the procedure, adapted from the literature [
28,
51]. Thus, the measurement of HPMo in the washing solvent in the Soxhlet extractor also made it possible to indirectly estimate the amount of HPMo immobilized efficiently by ultraviolet-visible (UV-vis) spectroscopy [
28,
51]. At the end of this period, the solid was recovered by filtration, washed extensively with water at room temperature and posteriorly characterized.
2.3. Characterization
The final chemical composition of the calcined samples was determined using a Shimadzu EDX-700 energy dispersive X-ray spectrometer (EDX; EDX-700, SHIMADZU, Kyoto, Japan), with a rhodium X-ray source tube (40 kV, SHIMADZU, Kyoto, Japan). The spectra were obtained using 500 mg of the powder material, deposited in a bottom sample holder made of polyethylene film with low X-ray absorption, in the range of work.
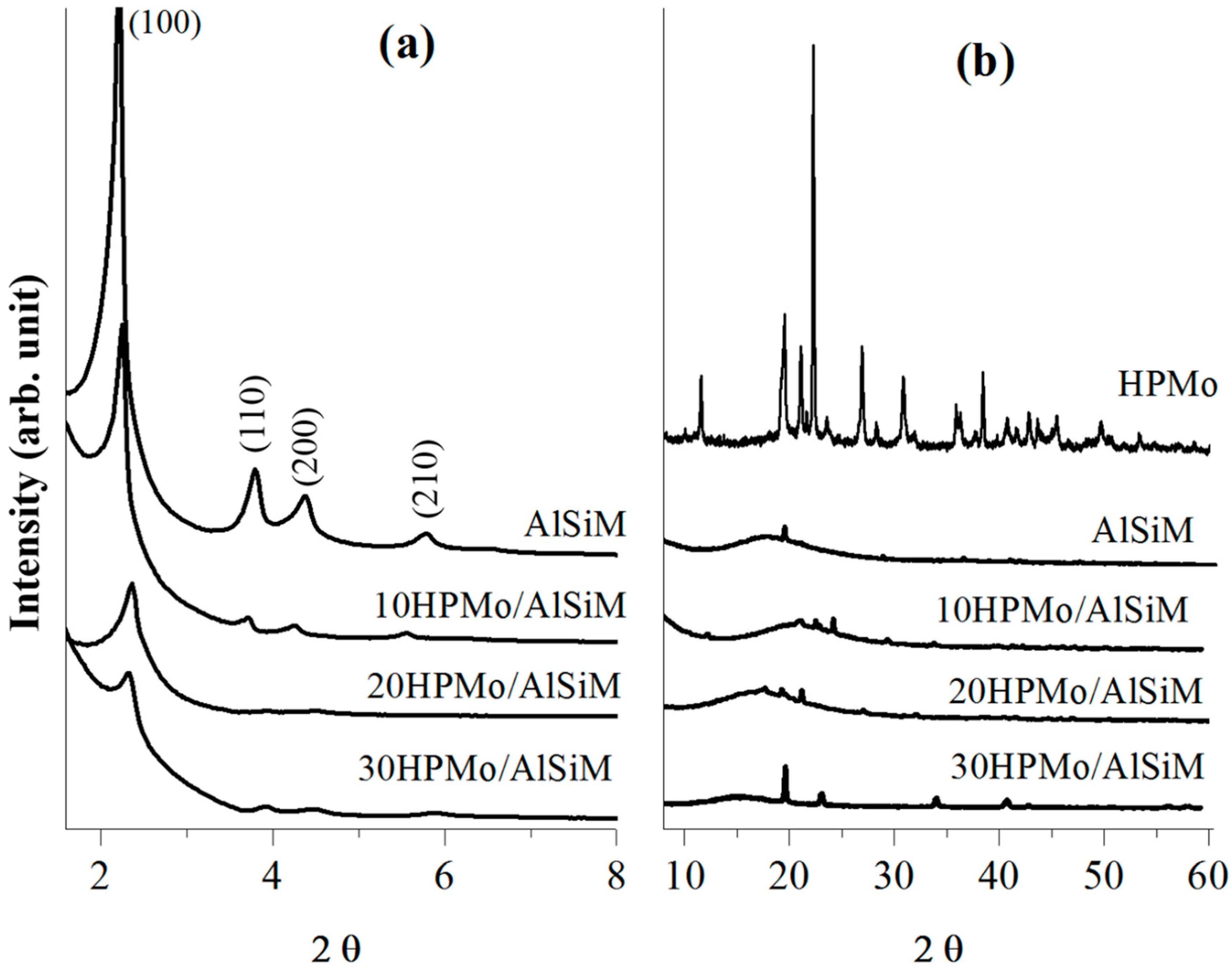
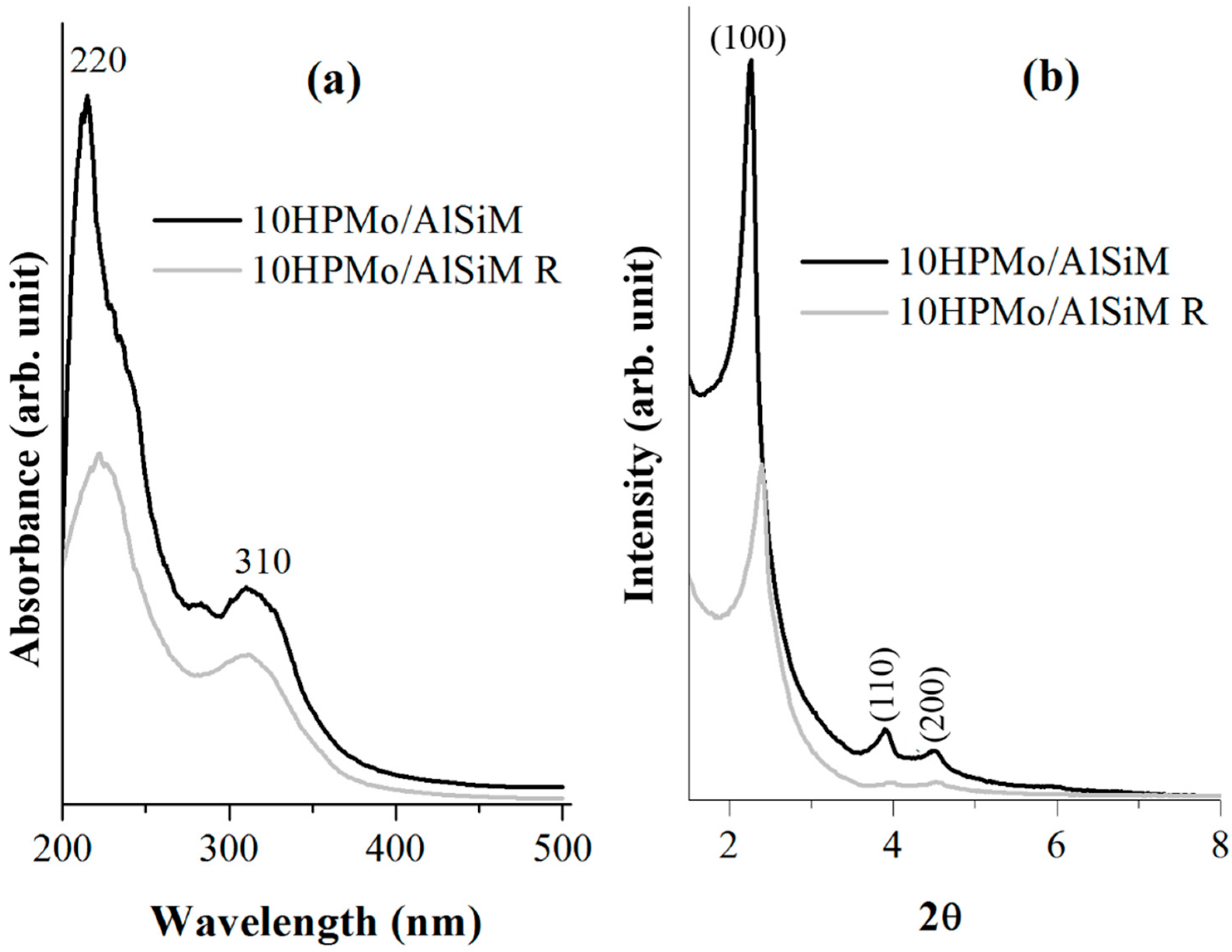
X-ray diffractograms were obtained on a Bruker D8 Advance diffractometer (Bruker D8 Advance; Bruker Corp, Billerica, MA, EUA), using the powder method, at a 1° < 2θ <10° interval. Cu Kα (λ = 1.5406 Å, 40 kV e 40 mA) radiation was used. The 2θ scanning speed was 0.02° min
−1. The distance (a
0) between pore centers of the hexagonal structure was calculated by means of the equation a
0 = 2
d100/√3 [
40].
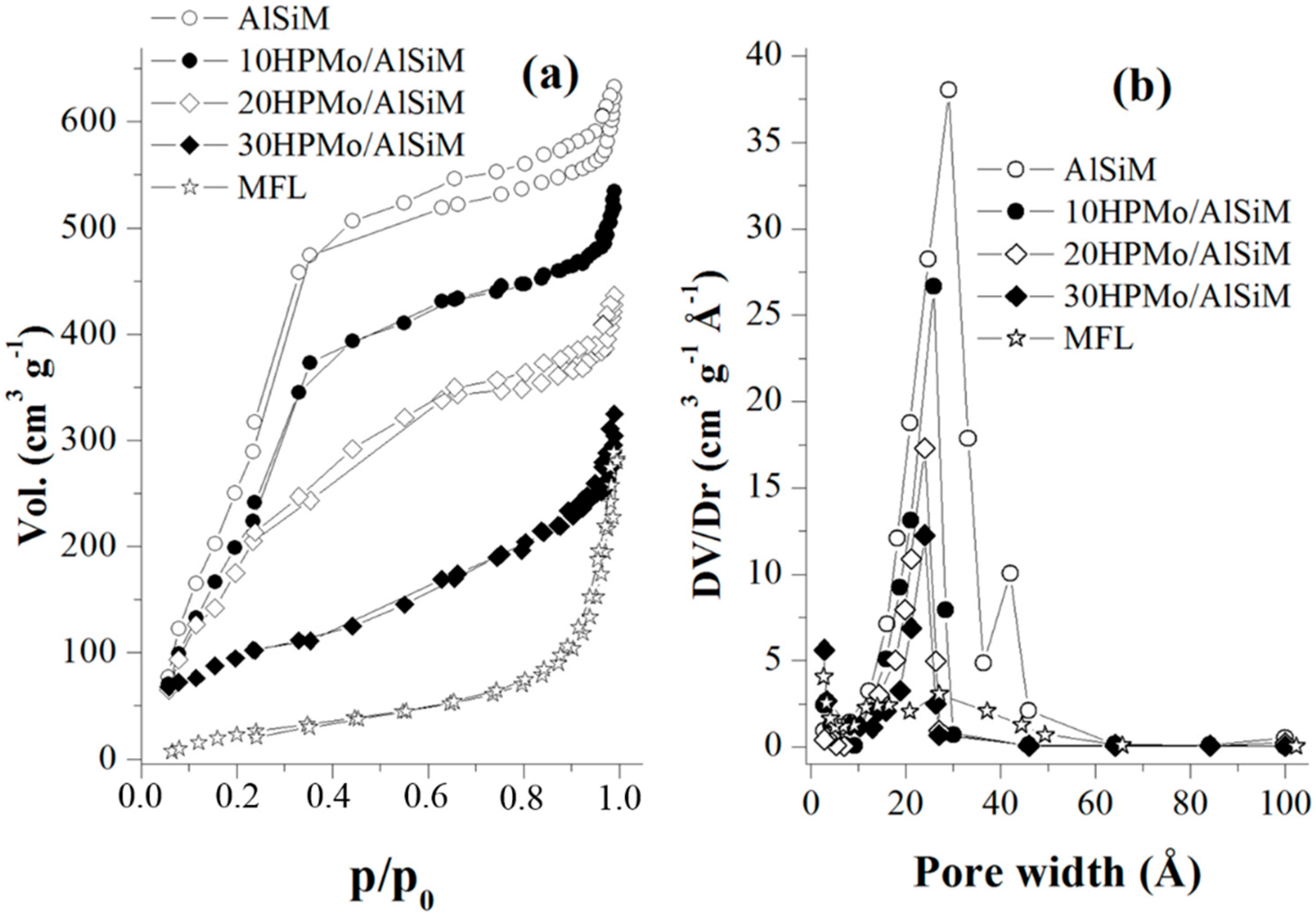
N
2 adsorption-desorption isotherms were obtained at liquid nitrogen temperature using a Micromeritics TriStar II model 3020 V1.03 apparatus (Micromeritics, Norcross, GA, USA). Before each measurement, the samples were outgassed at 200 °C for 2 h. The specific surface area (SSA) was determined according to the standard Brunauer-Emmett-Teller (BET) method. Pore diameter (D
p) and pore volume (V
p) were obtained by the Barret-Joyner-Halenda (BJH) method. The pore wall thickness (W
t) was calculated according to Equation W
t = a
0 − D
p [
40].
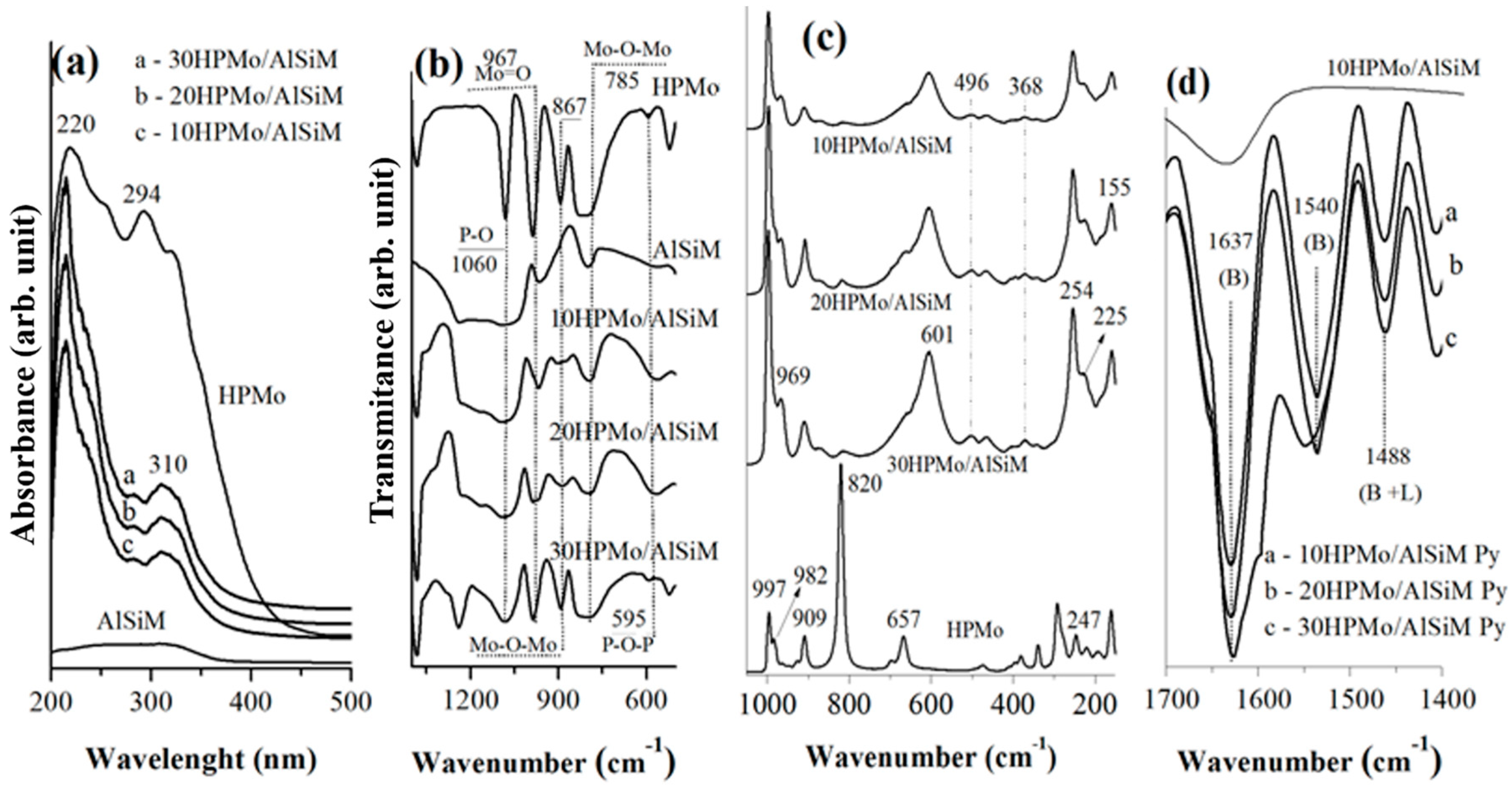
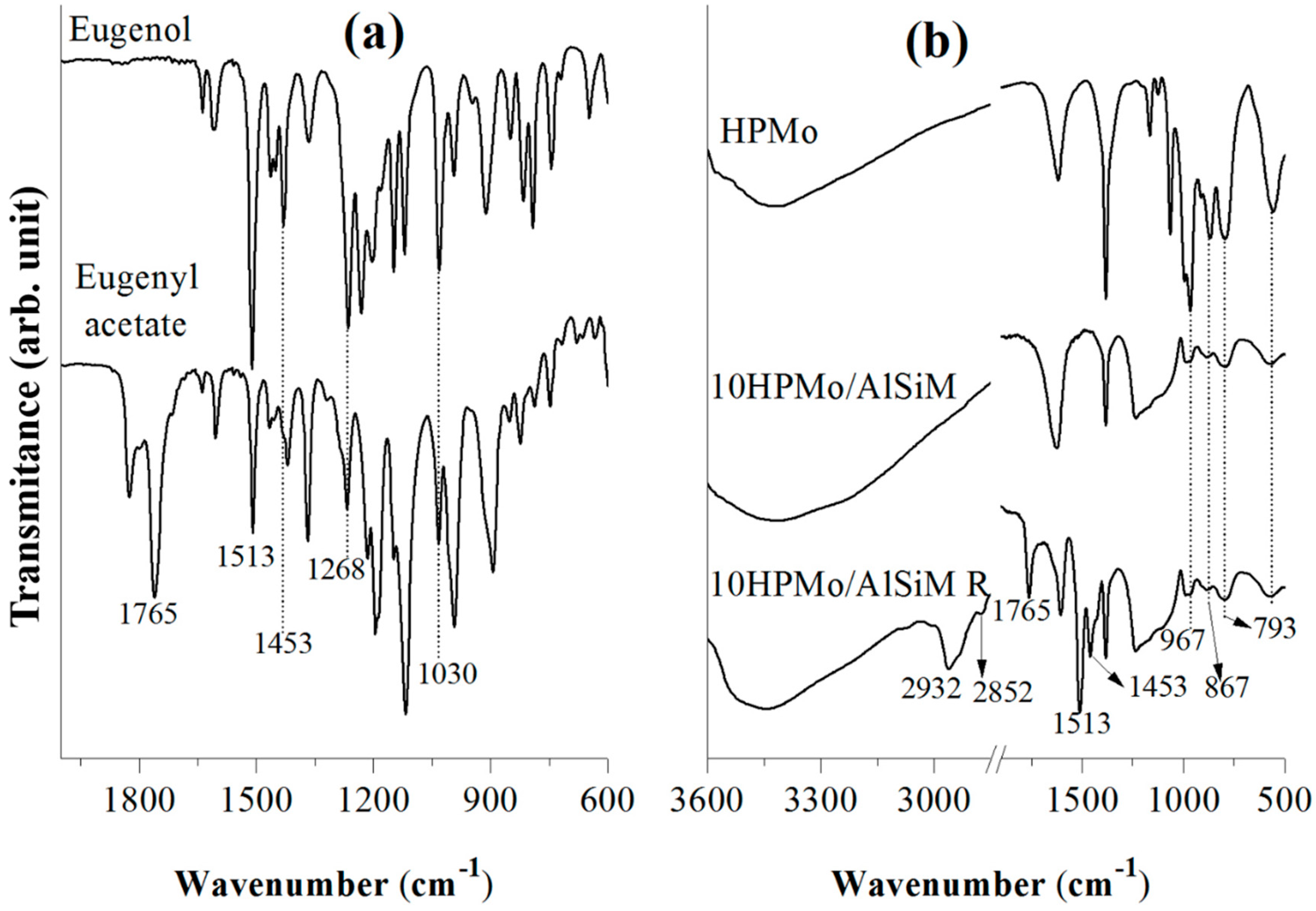
Fourier transform infrared spectroscopy (FTIR) spectra were obtained from a spectrophotometer of Shimadzu (Kyoto, Japan) model IRPrestige-21A with a resolution of 32 and 100 scans and analyzed by Thermo Electron Corporation, IR 100 model with a resolution of 4 and 32 scans. For the analysis of all materials KBr pellets were used and the spectra were obtained in the region 4000–400 cm−1.
Diffuse ultraviolet-visible reflectance spectroscopy (DRS) were recorded, in the range of 200–500 nm, on a Shuimadzu UV-vis model ISR-2600 Plus spectrophotometer (EDX; EDX-700, SHIMADZU, Kyoto, Japan).
The Raman spectra were recorded using a Bruker, Vertex 70V spectrometer (Bruker D8 Advance; Bruker Corp, Billerica, MA, EUA), the system using a Diode Pumped Solid State (DPSS) laser by an Nd: YAG with an excitation laser equal to 514 nm. The detector is of germanium (Ge) kept under refrigeration of liquid nitrogen.
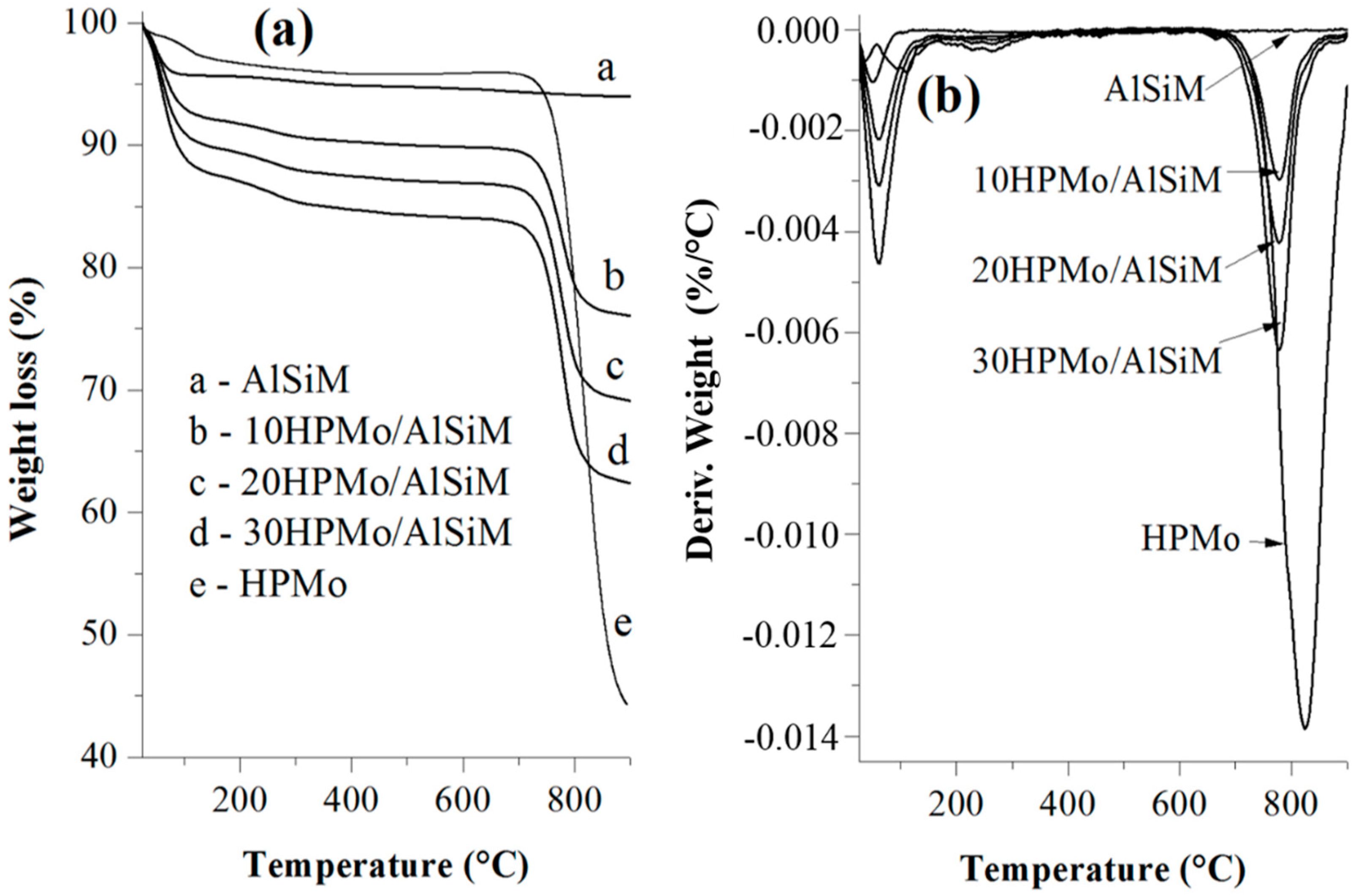
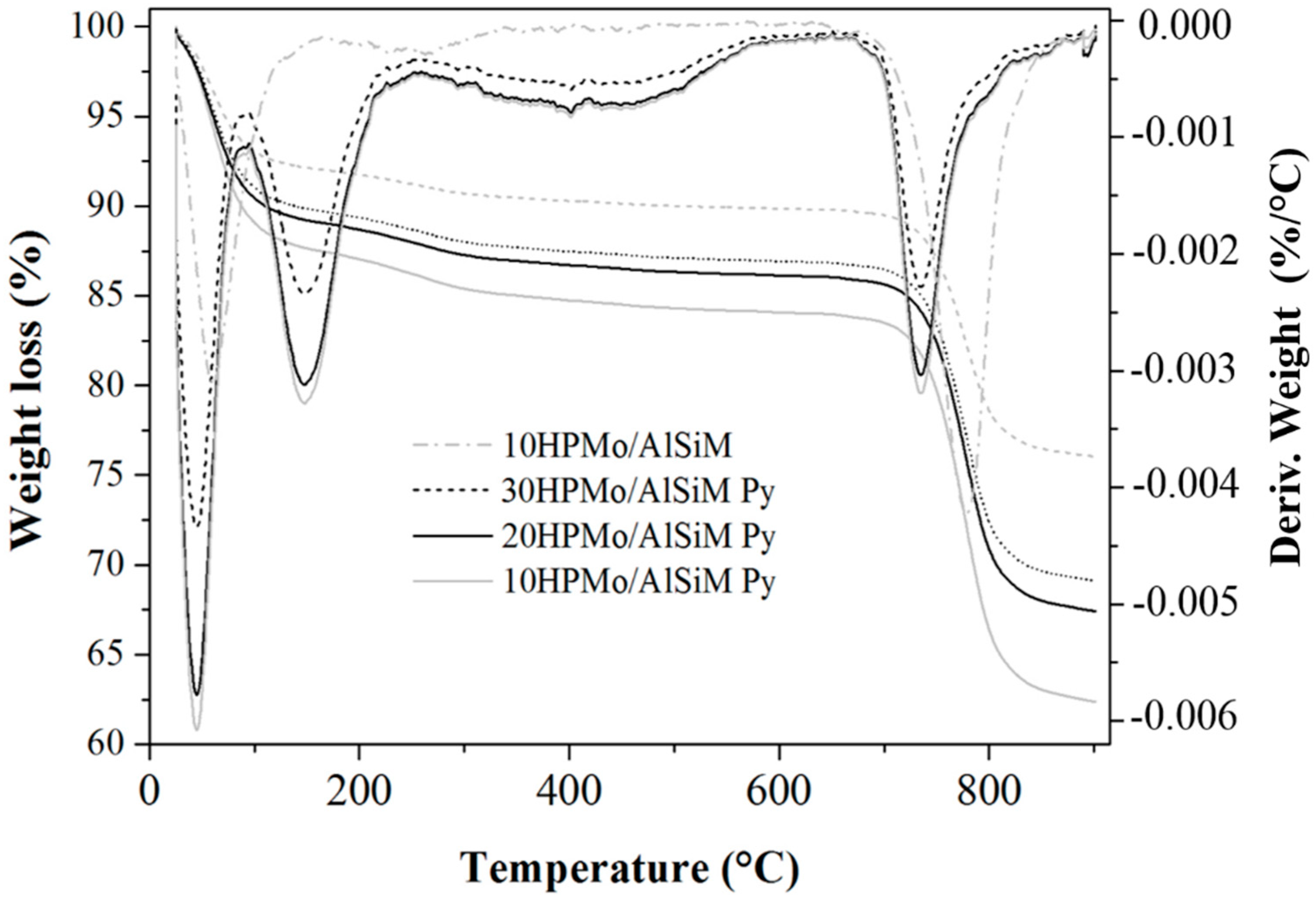
The thermogravimetric analysis (TGA/DTG) curves were obtained on a Shimadzu DTG-60H (Kyoto, Japan) model using N2 as purging gas (50 mL min−1). The analyses were performed from room temperature (~25 °C) to 900 °C at a rate of 10 °C min−1. About 10 mg of the samples were placed in platinum containers and heated from room temperature to 900 °C at a heating rate of 10 °C min−1 and using a N2 flow of 50 mL min−1.
The same technique was used for the quantification of the number of acid sites. For this, the samples prepared for evaluation of the presence of acidic sites in the infrared were submitted to thermal analysis and the TGA/DTG curves of the samples without adsorption and with adsorbed pyridine (Py) were evaluated. The number of acid sites was calculated from the difference in mass of the sample before and after being submitted to the adsorption of pyridine. The value of this difference corresponds to the mass of pyridine adsorbed where each mole of pyridine equals one mole of the acid site present on the surface of the catalyst. From these data the number of mmol of pyridine (nPy) per gram of sample was mathematically determined according to the method proposed by Nascimento et al. [
25,
29].
FTIR of adsorbed pyridine was the technique used to confirm the presence of Brønsted and Lewis acid centers in the catalysts [
28,
52]; 50 mg of sample was used for the acid sites identification that was made through previous heating at 120 °C during 90 min before the treatment with the pyridine probe molecule. After cooling, the sample were scaned within the range from 1700 and 1400 cm
−1 in the FTIR spectrum [
28,
52].
The surface acidity was determined using acid-base titration [
26,
28,
30]. In a typical measurement, 0.1 g of the solid was dispersed in 50 mL of 0.1 mol L
−1 NaCl. The dispersion was stirred for 24 h and titrated with 0.1 mol L
−1 NaOH in the presence of phenolphthalein.
Tests to determine the leaching of the HPMo of the catalysts were performed in UV-vis equipment of Thermo-scientific (Waltham, MA, USA), model Evolution array UV-vis spectrophotometer, with 200 to 600 nm scan and 30 scan resolution. The liquids were placed in a quartz tube. The calibration curve was constructed using equations (y = 0.1001x + 0.0032) of absorbance of λ
max = 310 nm with excellent correlation coefficient (R
2 = 0.9999). To quantify HPMo leached in the reaction medium, another analytical curve was constructed from the post-reaction solution (1 to 5 mg L
−1 of HPMo) which was appropriately diluted with 0.1 mol L
−1 HCl to avoid any hydrolysis of the anion [PMo
12O
40]
3−, with an equation (y = 0.0999 + 0.0025) absorbance of λ
max = 310 nm and an excellent correlation coefficient (R
2 = 0.9999) based on that described in the literature [
28,
51].
2.4. Catalytic Tests
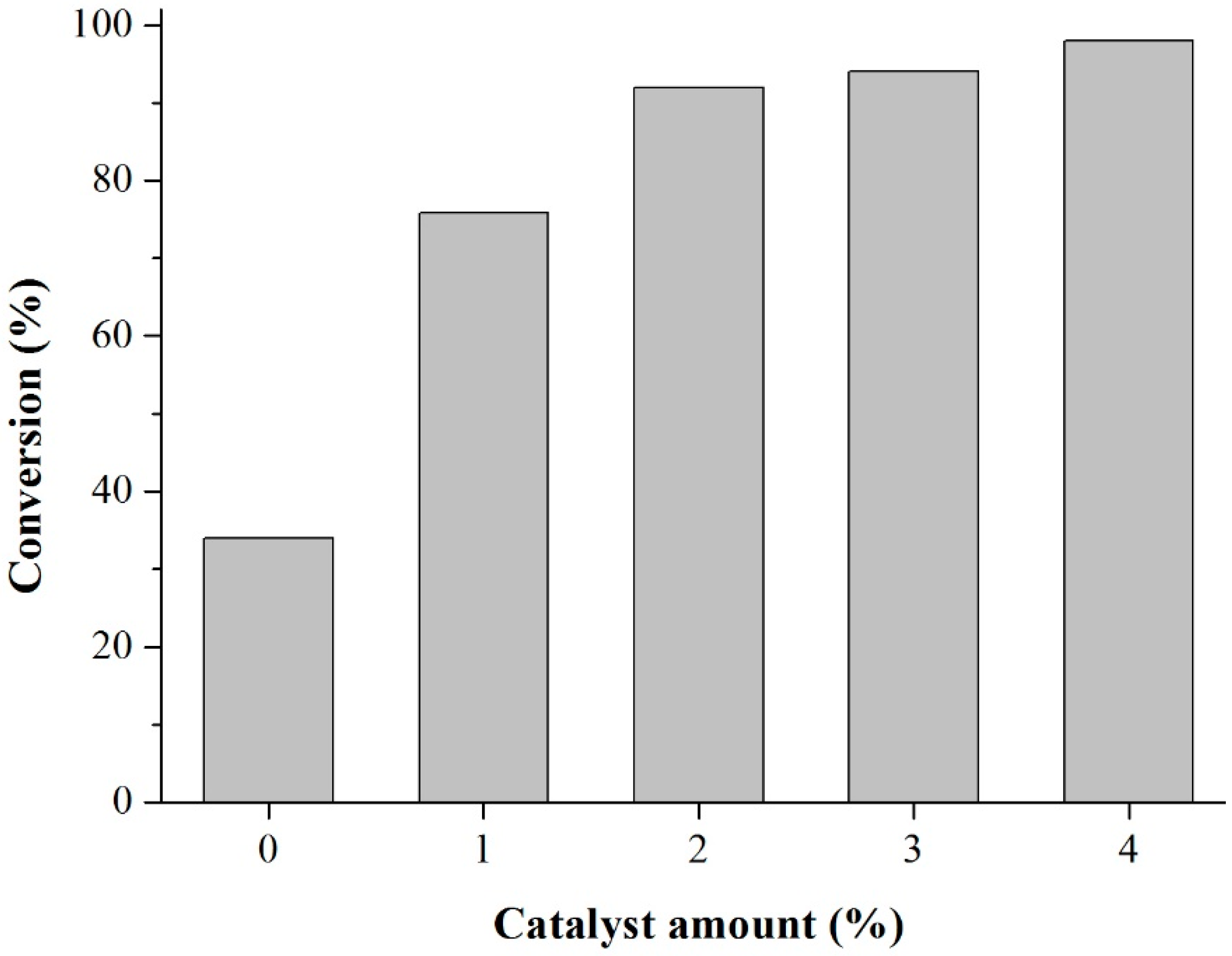
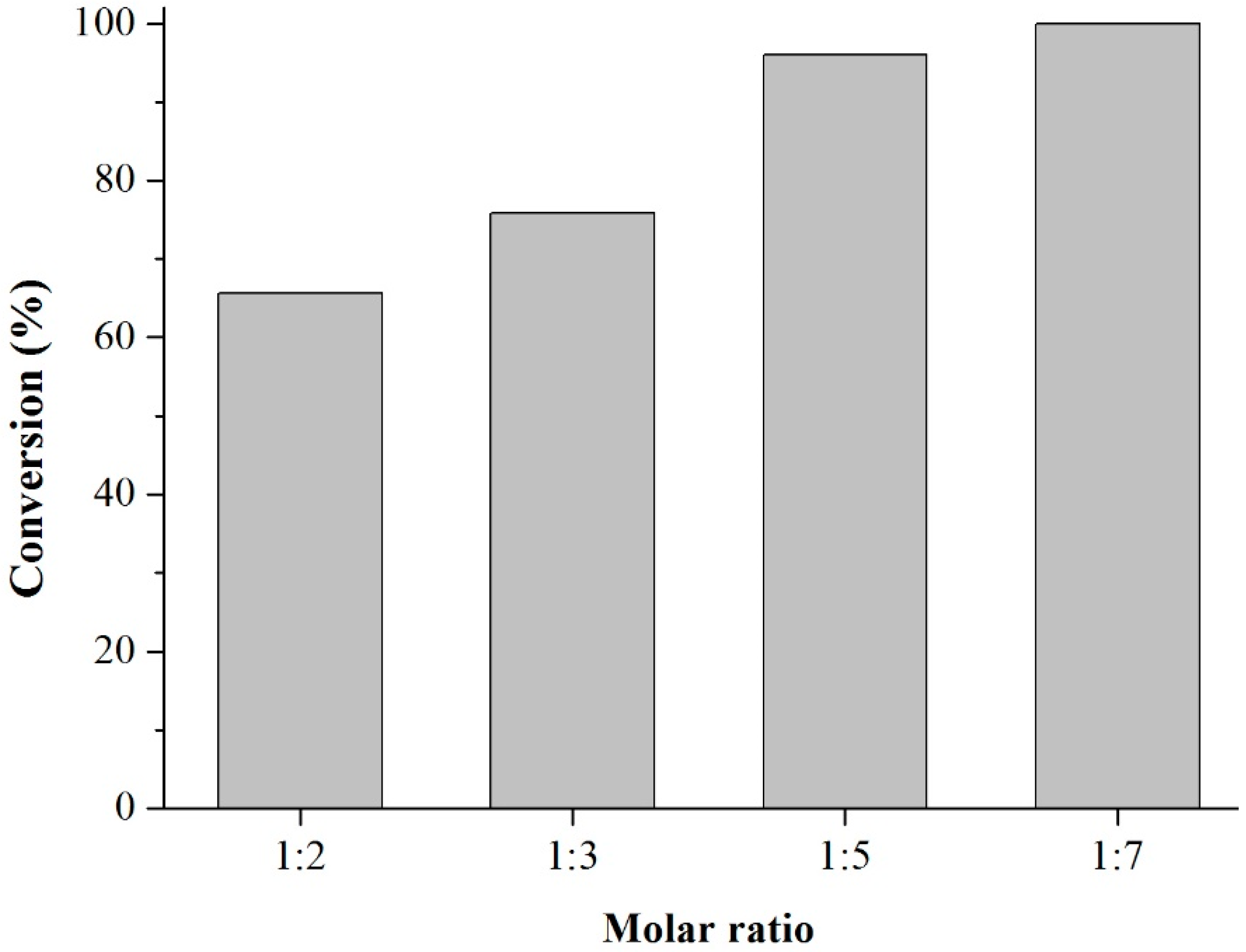
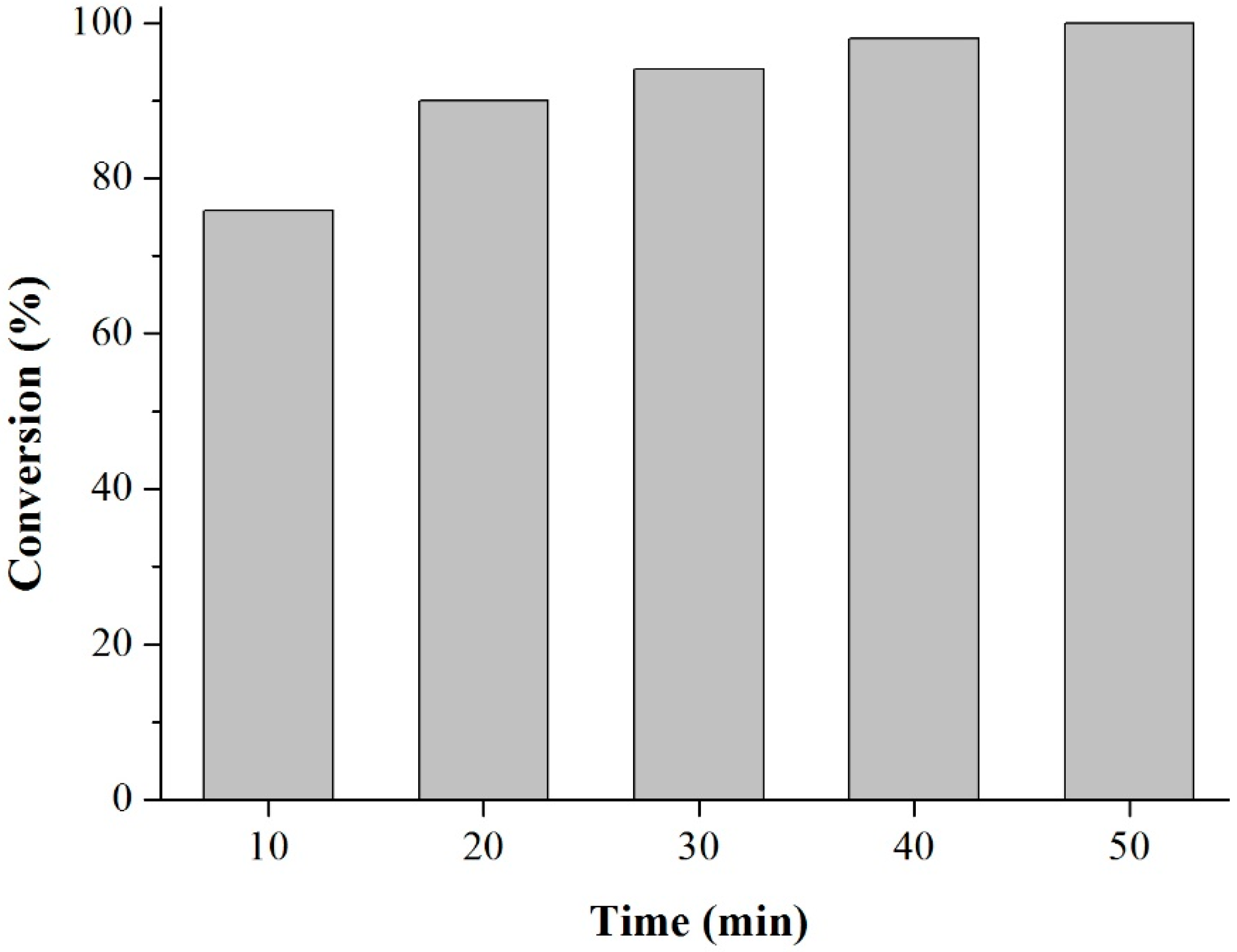
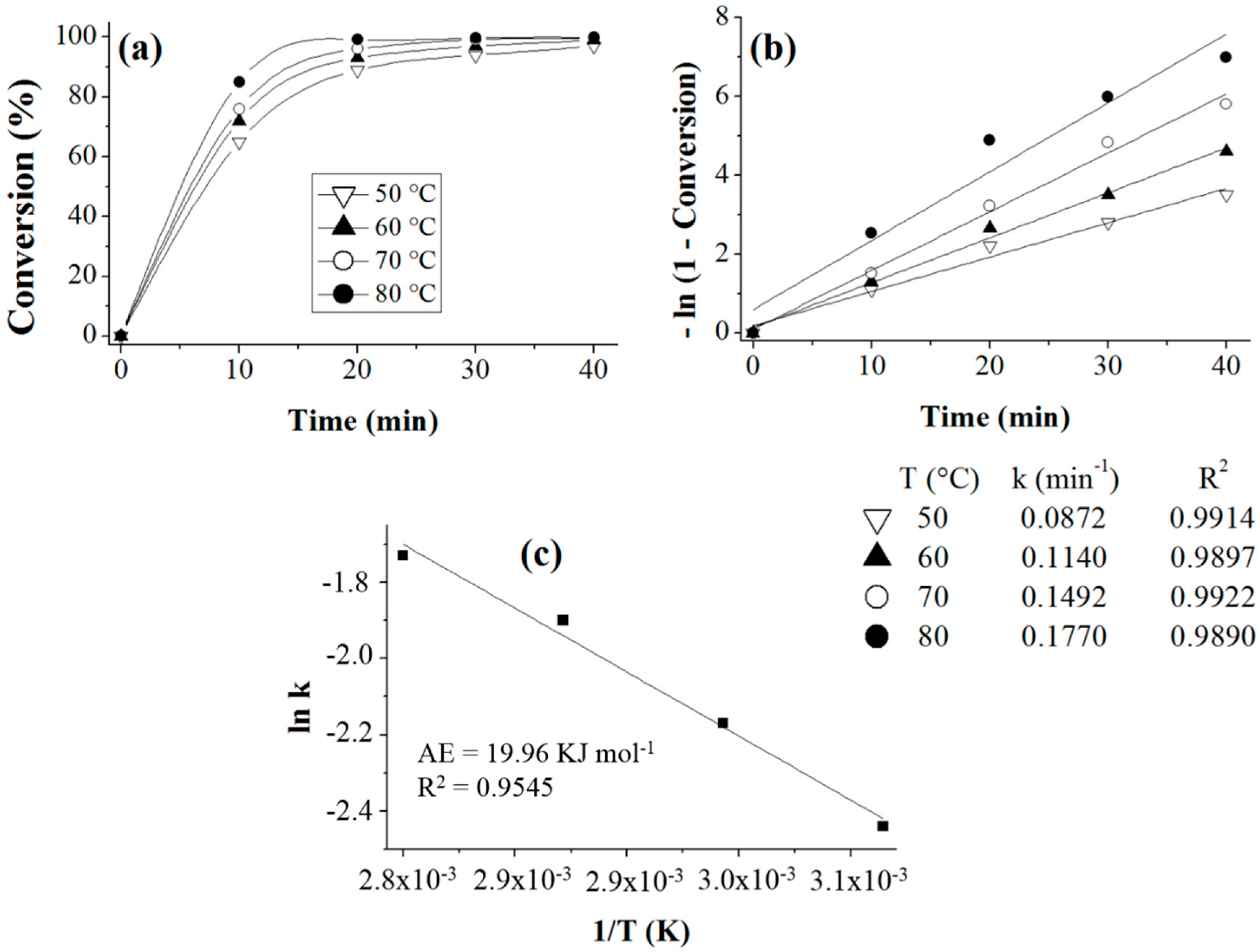
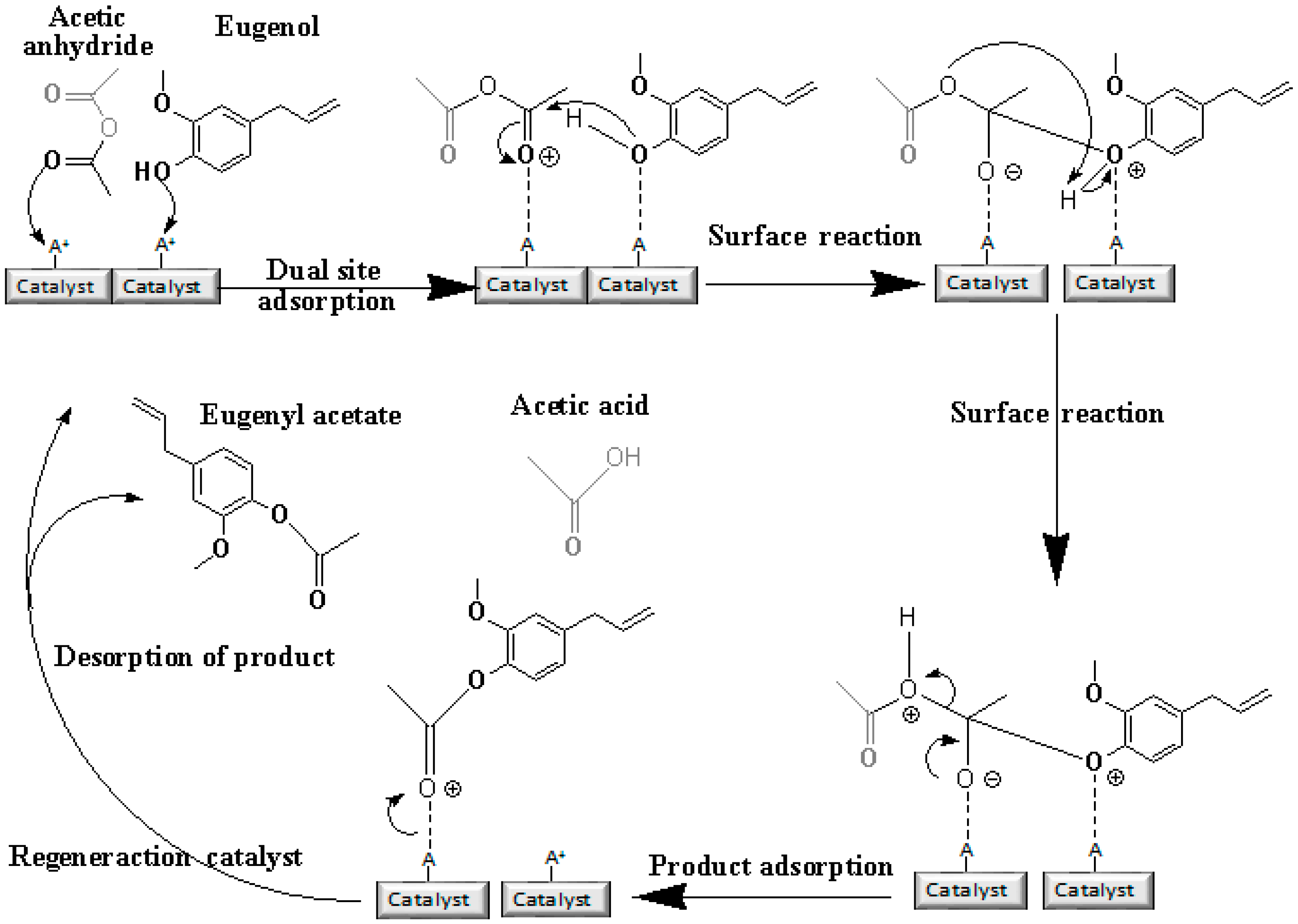
Acetylation of Eugenol
Prior to the experiments, the catalysts were activated at 130 °C for 2 h. Tests of the catalysts were conducted in one run on a multirreator PARR 4871 (Parr Instrument Company, Moline, IL, USA). In a typical experiment, the eugenol was mixed with acetic anhydride at the molar ratio of 1:5 (Eugenol: AA) and 2%
w/
w of the solid acid catalyst (related to the Eugenol weight). The reaction mixture was stirred (500 rpm) and warmed from room temperature to 80 °C. Once the desired temperature was reached, the system was maintained for 40 min (
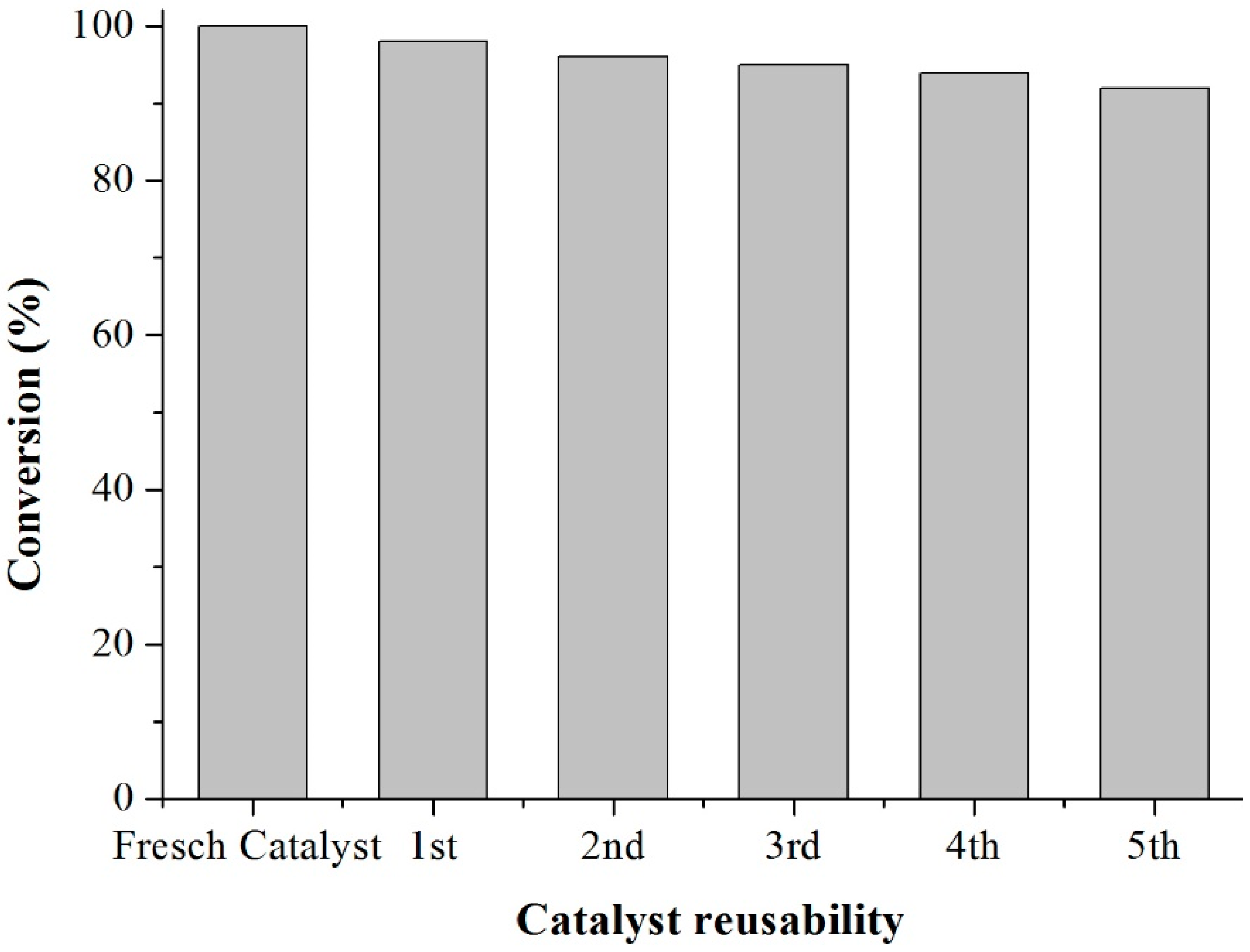
Scheme 3). This period was considered as the kinetic contact time. At the end of the reaction, the reaction mixture was separated by centrifugation at 6000 rpm for 10 min. The separated catalyst by centrifugation was washed with ethanol to remove the polar and non-polar organic compounds adsorbed on the catalyst surface and oven dried at 150 °C for 4 h before being reused in the next cycle with fresh reagents to study their reusability. The recycle study was conducted in a manner similar to the above, but the mass of the catalyst and the other reactants were always recalculated prior to use in the subsequent reaction cycles under the best reaction conditions (80 °C for 40 min).
The acetic anhydride and acetic acid present in the filtrate were removed by rotevaporation at 70 °C for 4 h. After this procedure, an aliquot (1 μL) of the product was diluted in 1.5 mL of dichloromethane and analyzed by gas chromatography. Qualitative and quantitative analysis was performed through gas chromatography-mass spectrometry (GC-MS, Shimadzu QP2010 plus instrument, Shimadzu Corporation, Kyoto, Japan) under the following conditions: silica capillary column Rtx-5MS (30 m × 0.25 mm × 0.25 mm film thickness, Shimadzu Corporation, Kyoto, Japan); programmed temperature, 60–240 °C (3 °C min
−1); temperature of the injector, 200 °C; carrier gas, helium, adjusted at a linear velocity of 1.2 mL min
−1; type of injection, without division; the divided stream was adjusted to yield a ratio of 20:1; septal scan was a constant 10 mL min
−1; EIMS (Electron Ionization Mass Spectra), electronic energy, 70 eV; temperature of the source of ions and bonding parts, 200 °C. The retention index was calculated for all volatile constituents using a homologous series of n-alkanes (C8–C32, Sigma-Aldrich, Saint Louis Missouri, EUA) The constituents were identified by comparing their mass spectra and retention indices (RI) with those in the system library (NIST-11, FFNSC-2) [
53] and in the literature [
54,
55].

 ,
, 


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}