
Abstract
Structures and energies of XH4 + and XH6 + (X = B, Al and Ga) have been calculated at the density functional theory (DFT) B3LYP/6-311++G(3df,2pd) level. Calculations indicate that although the structure with a three center two electron (3c-2e) bond is the global minimum for BH4 +, the global minima of AlH4 + and GaH4 + are not those with one 3c-2e bond, but those with two 3c-2e bonds. For calibration, both structures of AlH4 + were also calculated at the ab initio CCSD(T)/cc-pVTZ level and results in agreement with the DFT results were found. Similar calculations also indicate that although the C2v symmetrical structure with two 3c-2e bonds is the global minimum for BH6 +, the global minima of AlH6 + and GaH6 + are not the C2v symmetrical structures with two 3c-2e bonds but the C2 symmetrical structures with three 3c-2e bonds.
Similar content being viewed by others
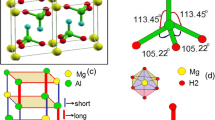

Author information
Authors and Affiliations
Electronic Supplementary Material
Rights and permissions
About this article
Cite this article
Salzbrunn, S., Rasul, G., Surya Prakash, G. et al. Structures of XH4 + and XH6 + (X = B, Al and Ga) Cations. J Mol Model 6, 213–216 (2000). https://doi.org/10.1007/s0089400060213
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s0089400060213