
Abstract
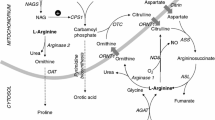
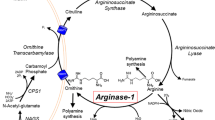
Deficiency of liver arginase (AI) is characterized clinically by hyperargininemia, progressive mental impairment, growth retardation, spasticity, and periodic episodes of hyperammonemia. The rarest of the inborn errors of urea cycle enzymes, it has been considered the least life-threatening, by virtue of the typical absence of catastrophic neonatal hyperammonemia and its compatibility with a longer life span. This has been attributed to the persistence of some ureagenesis in these patients through the activity of a second isozyme of arginase (AII) located predominantly in the kidney. We have treated a number of arginase-deficient patients into young adulthood. While they are severely retarded and wheelchairbound, their general medical care has been quite tractable. Recently, however, two of the oldest (M.U., age 20, and M.O., age 22) underwent rapid deterioration, ending in hyperammonemic coma and death, precipitated by relatively minor viral respiratory illnesses inducing a catabolic state with increased endogenous nitrogen load. In both cases, postmortem examination revealed severe global cerebral edema and aspiration pneumonia. Enzyme assays confirmed the absence of AI activity in the livers of both patients. In contrast, AII activity (identified by its different cation cofactor requirements and lack of precipitation with anti-AI antibody) was markedly elevated in kidney tissues, 20-fold in M.O. and 34-fold in M.U. Terminal plasma arginine (1500μmol/1) and ammonia (1693 mmol/1) levels of M.U. were substantially higher than those of M.O. (348μmol/1 and 259μmol/1, respectively). By Northern blot analysis, AI mRNA was detected in M.O.'s liver but not in M.U.'s; similarly, anti-AI crossreacting material was observed by Western blot in M.O. only. These findings indicate that, despite their more longlived course, patients with arginase deficiency remain vulnerable to the same catastrophic events of hyperammonemia that patients with other urea cycle disorders typically suffer in infancy. Further, unlike those other disorders, an attempt is made to compensate for the primary enzyme deficiency by induction of another isozyme in a different tissue. Such substrate-stimulated induction of an enzyme may be unique in a medical genetics setting and raises novel options for eventual gene therapy of this disorder.
Similar content being viewed by others
References
Batshaw ML, Busilow S, Waber L, Blom W, Brubakk AM, Burton BK, Cann HM, Kerr D, Mamunes P, Matalon R, Myerberg D, Schafer IA (1982) Treatment of inborn errors of urea synthesis: activation of alternative pathways of waste nitrogen synthesis and excretion. N Engl J Med 306:1387–1392
Bernar J, Hanson RA, Kern R, Phoenix B, Shaw KNF, Cederbaum SD (1986) Arginase deficiency in a 12-year-old boy with mild impairment of intellectual function. J Pediatr 108:432–435
Brusilow SW, Horwich AL (1989) Urea cycle enzymes. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The metabolic basis of inherited disease, 6th edn. McGraw-Hill, New York, pp 629–663
Carvajal N, Cederbaum SD (1986) Kinetics of inhibition of rat liver and kidney arginase by proline and branched-chain amino acids. Biochim Biophys Acta 870:181–184
Cederbaum SD, Shaw KNF, Valente M (1977) Hyperargininemia. J Pediatr 90:569–573
Cederbaum SD, Shaw KNF, Spector EB, Verity MA, Snodgrass PJ, Sugarman GI (1979) Hyperargininemia with arginase deficiency. Pediatr Res 13:827–833
Cederbaum SD, Moedjono SJ, Shaw KNF, Carter M, Naylor E, Walser M (1982) Treatment of hyperargininemia due to arginase deficiency with a chemically defined diet. J Inherited Metab Dis 5:95–99
Chirgwin JJ, Przbyla AE, MacDonald RJ, Rutter WJ (1979) Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochemistry 18:5294–5299
Dizikes GJ, Grody WW, Kern RM, Cederbaum SD (1986) Isolation of human liver arginase cDNA and demonstration of nonhomology between the two human arginase genes. Biochem Biophys Res Commun 141:53–59
Gahl WA, Kaiser-Kupfer MI (1987) Complications of nephropathic cystinosis after renal failure. Pediatr Nephrol 1:260–268
Gasiorowska I, Porembska Z, Jachimowicz J, Mochnaka I (1970) Isozymes of arginase in rat tissues. Acta Biochim Pol 17:19–30
Gatti RA, Concannon P, Salser W (1984) Multiple use of Southern blots. Bio Techniques 2:148–155
Goldberg DA (1980) Isolation and partial characterization of the Drosophila alcohol dehydrogenase gene. Proc Natl Acad Sci USA 77:5794–5798
Grody WW, Dizikes GJ, Cederbaum SD (1986) Human arginase isozymes. In: Rattazzi MC, Scandalios JG, Whitt GS (eds) Isozymes: current topics in biological and medical research. Liss, New York, pp 181–214
Grody WW, Argyle C, Kern RM, Dizikes GJ, Spector EB, Strickland AD, Klein D, Cederbaum SD (1989) Differential expression of the two human arginase genes in hyperargininemia: enzymatic, pathologic, and molecular analysis. J Clin Invest 83:602–609
Grody WW, Klein D, Dodson AE, Kern RM, Wissmann PB, Goodman BK, Bassand P, Marescau B, Kang S-S, Leonard JV, Cederbaum SD (1992) Molecular genetic study of human arginase deficiency. Am J Hum Genet 50:1281–1290
Hammond JE, Savory J (1976) Advances in the detection of amino acids in biological fluids. Ann Clin Lab Sci 6:158–166
Heidenreich R, Natowicz M, Hainline BE, Berman P, Kelley RI, Hillman RE, Berry GT (1988) Acute extrapyramidal syndrome in methylmalonic acidemia: “metabolic stroke” involving the globus pallidus. J Pediatr 113:1022–1027
Herzfeld A, Raper SM (1976) The heterogeneity of arginases in rat tissues. Biochem J 153:469–478
Kaiser-Kupfer MI, Caruso RC, Minkler DS, Gahl WA (1986) Longterm ocular manifestations in nephropathic cystinosis. Arch Ophthalmol 104:706–711
Kaufman FR, Kogut MD, Donnell GN, Goebelsmann U, March C, Koch R (1981) Hypergonadotropic hypogonadism in female patients with galactosemia. N Engl J Med 304:994–998
Kunkel LM, Smith KD, Boyer SH, Borgaonkar DS, Wachtel SS, Miller OJ, Breg WR, Jones HW, Rary JM (1977) Analysis of human Y-chromosome-specific reiterated DNA in chromosome variants. Proc Natl Acad Sci USA 74:1245–1249
Lehrach H, Diamond D, Wozney JM, Boedtker H (1977) RNA molecular weight determinations by gel electrophoresis under denaturing conditions: a critical reexamination. Biochemistry 16:4743
Lenke RR, Levy HL (1980) Maternal phenylketonuria and hyperphenylalaninemia. N Engl J Med 303:1202–1208
Michels W, Beaudet AL (1978) Arginase deficiency in multiple tissues in argininemia. Clin Genet 13:61–67
Schimke RT (1964) Enzymes of arginine metabolism in mammalian cell culture. I. Repression of argininosuccinate synthetase and argininosuccinase. J Biol Chem 239:136–144
Spector EB, Keirnan M, Bernard B, Cederbaum SD (1980) Properties of fetal and adult red blood cell arginase: a possible prenatal diagnostic test for arginase deficiency. Am J Hum Genet 32:79–87
Spector EB, Rice SCH, Moedjono S, Bernard B, Cederbaum SD (1982) Biochemical properties of arginase in human adult and fetal tissues. Biochem Med 28:165–175
Spector EB, Rice SCH, Cederbaum SD (1983) Immunologic studies of arginase in tissues of normal human adult and arginase-deficient patients. Pediatr Res 17:941–944
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Grody, W.W., Kern, R.M., Klein, D. et al. Arginase deficiency manifesting delayed clinical sequelae and induction of a kidney arginase isozyme. Hum Genet 91, 1–5 (1993). https://doi.org/10.1007/BF00230212
Received:
Revised:
Issue Date:
DOI: https://doi.org/10.1007/BF00230212