
Abstract
A large volume of biological data is being generated for studying mechanisms of various biological processes. These precious data enable large-scale computational analyses to gain biological insights. However, it remains a challenge to mine the data efficiently for knowledge discovery. The heterogeneity of these data makes it difficult to consistently integrate them, slowing down the process of biological discovery. We introduce a data processing paradigm to identify key factors in biological processes via systematic collection of gene expression datasets, primary analysis of data, and evaluation of consistent signals. To demonstrate its effectiveness, our paradigm was applied to epidermal development and identified many genes that play a potential role in this process. Besides the known epidermal development genes, a substantial proportion of the identified genes are still not supported by gain- or loss-of-function studies, yielding many novel genes for future studies. Among them, we selected a top gene for loss-of-function experimental validation and confirmed its function in epidermal differentiation, proving the ability of this paradigm to identify new factors in biological processes. In addition, this paradigm revealed many key genes in cold-induced thermogenesis using data from cold-challenged tissues, demonstrating its generalizability. This paradigm can lead to fruitful results for studying molecular mechanisms in an era of explosive accumulation of publicly available biological data.
Similar content being viewed by others


Introduction
The huge amount of data generated from previous biological studies provides a precious resource for mining new biological knowledge. A significant portion of the data is freely available in public repositories such as ArrayExpress1 and Gene Expression Omnibus (GEO)2. For example, around one million series studies are publicly available in GEO. Due to the unstructured nature of the metadata associated with public data, manual curation is required3,4,5,6,7, a step that is essential for collecting large-scale gene expression data.
Gene expression data facilitate the application of the network reconstruction approach for identifying key factors in biological processes. For example, Bhaduri et al.8 applied the gene network reconstruction approach to explore epidermal differentiation regulators. Using network analysis, the MPZL3 gene was identified as a highly connected hub required for epidermal differentiation. In addition, the MPZL3 gene indirectly regulates epidermis genes, including ZNF750, TP63, KLF4, and RCOR1, through the FDXR gene and reactive oxygen species.
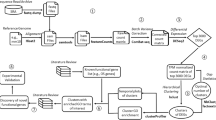
Complementing data analyses with more relevant data improves the identification of key factors in biological processes. Even though massive expression data can provide essential insights in revealing genetic interactions, there are confounding factors or “noise” introduced by technical variations, such as batch effects9. To obviate the “noise” and generate a consistent result, one solution is integrative analysis by comparing large-scale datasets10. In this report, we introduce a paradigm to integrate data collection and data analysis for mining key factors in specific biological processes (Fig. 1). To demonstrate the power of our data processing paradigm, we evaluate key factors of two applications in skin biology and energy homeostasis.

Data processing paradigm flowchart. Data curation was performed to identify the gene expression datasets with the given biological process perturbed (e.g., the process is increased in CMP 1 with \(+1\) and is decreased in CMP 2 or CMP m with direction \(-1\)). DEG analysis was performed on the curated datasets, and \(+1/-1/0\) represents the up-regulated, down-regulated, or unchanged genes, respectively. To prioritize important genes in the biological process for each gene in a curation dataset, an affinity score of \(+1/-1/0\) was calculated first by comparing the gene expression change and the regulation of the biological process, where \(+1\) indicates that the gene (e.g., Gene 1 in CMP 1 and CMP 2) is positively related to the biological process, \(-1\) indicates that the gene (e.g., Gene 2 in CMP m and Gene n in CMP 1) is negatively related to the biological process, and \(0\) indicates no relation of the gene to the biological process. No measurement (notated as NA, e.g., Gene 3 in CMP 1) indicates an unknown affinity of the gene in the dataset. By summing the affinity scores, a consensus score was calculated for genes in the perturbed datasets. Genes with higher consensus scores were identified as more related to the biological process.
The epidermis of skin mediates various functions that protect against the environment, such as microbial pathogen challenges, oxidant stress, ultraviolet light, chemicals, and mechanical insults11. Therefore, it is critical to understand mechanisms of epidermal development to develop new treatment for human skin diseases12. Our paradigm predicts key factors in epidermal development by collecting related datasets and integrating the information. A fraction of genes are annotated in Gene Ontology (GO) or have strong functional validation based on gain-/loss-of-function studies13. The remaining genes are novel; their functionality has not been experimentally validated. We picked a top hit, suprabasin (SBSN), and performed loss-of-function experiments for the mouse homolog of gene Sbsn using RNA-Seq. The analysis validates that Sbsn knockdown in mouse keratinocyte cultures down-regulates cornified envelope genes, suggesting an essential role of SBSN in epidermal differentiation. These results demonstrate the effectiveness of our paradigm in discovering key factors of epidermal development.
As another application, cold-induced thermogenesis (CIT) can reduce body weight by increasing resting energy expenditure in mammals14. Genes involved in CIT can be promising therapeutic targets for treating obesity and diabetes. Thus, it is important to understand the underlying mechanism of CIT. Our paradigm detected potential CIT-related genes, including known CIT genes and novel ones, showing that the paradigm can be generalized easily to other biological processes. It is a promising integrative analysis approach to identify key factors in biological processes.
Results
Identification of candidate epidermal development genes
To identify key gene expression datasets that are likely to be related to epidermal development, data curation was performed. A total of 295 epidermis development genes (according to GO) were searched on ArrayExpress to query microarray datasets, and over 300 datasets were retrieved. Due to the limitation of the search function in ArrayExpress, many retrieved datasets did not have any perturbation of these epidermis development genes, even though the gene symbols were mentioned in the datasets. To overcome this problem, manual curation was performed on each retrieved dataset to retain relevant ones, and the manual curation resulted in 24 experimental comparisons from 17 datasets with gain or loss function of 14 epidermis development genes (Table S1 and Methods).
To determine the candidate genes potentially involved in epidermal development, differential gene expression (DEG) analysis was performed on the 24 experimental comparisons of the curated microarray datasets. Differentially expressed genes were identified under \(q\le 0.05\). The large-scale gene expression changes derived from our curated datasets provided a list of candidate genes that may be potentially involved in epidermal development (Fig. 2).

DEG results of the curated microarray datasets. To identify the differentially expressed genes of the 24 experimental comparisons in curated microarray datasets, DEG analysis was performed as mentioned in supplemental materials. DEGs were identified under \(q\le 0.05\). (a) The bar plot depicts the number of DEGs identified in each of the 24 experimental comparisons. (b) The figure depicts the number of genes differentially expressed in n comparisons out of all the 24 comparisons. A large number of DEGs were identified in the curated datasets. A small group of genes was differentially expressed in multiple datasets.
To identify genes that are potentially critical in epidermal development, consensus gene scores were summarized for each gene from affinities on the 24 experimental comparisons. Eighty-one genes were identified as key genes related to epidermal development with a consensus score \(\ge 6\) (Table S2). The heatmap (Fig. S1) shows a majority of these genes with a \(+1\) affinity score in skin-related cell types. This information suggests that these top genes may play a role in epidermal development. To infer the biological processes involved, GO analysis was performed on these top genes using Fisher’s exact test (the null hypothesis is log-odds-ratio \( < 2\)) with all the genes annotated in GO as the background. Several epidermis-related GO terms were enriched in these genes (Fig. 3). For example, the essential GO terms in the epidermis were enriched, such as keratinocyte differentiation, epidermal cell differentiation, epidermis development, skin development, cornified envelope, and keratinization. In addition, the GO terms involved in skin barrier formation were also enriched, such as fatty acid elongase activity, lipoxygenase pathway, and establishment of skin barrier. These enriched GO terms suggest that the top identified genes are critical in epidermal development.

Biological process and literature study of genes with consensus score \(\ge 6\). To identify the biological process that the 81 top genes (consensus score \(\ge 6\)) were involved in, a GO enrichment analysis was performed. The enriched GO pathways were plotted with a log10 p-value, along with their log10 odds ratios in the enrichment analysis.
Because GO annotation is not complete for gene functions15, we manually curated functional annotations for the top identified genes. Of these genes, besides the 18 genes annotated in the GO term “epidermis development,” only three genes have loss-of-function experiments supporting their role in epidermal development. However, the majority of these identified genes have no functional experimental validation on epidermal development. Of the three genes with literature evidence, EDN1 (consensus score = 7) mediates the homeostasis of melanocyte (located at the bottom of epidermis) in vivo upon ultraviolet irradiation16. The loss function of ELOVL4 (consensus score = 6) represses the generation of very-long-chain fatty acids, which is critical for the epidermal barrier function, showing the important role of ELOVL4 in epidermis development17. The in vitro loss-of-function experiment of HOPX (consensus score = 6) leads to increased expression of cell differentiation markers in human keratinocytes, demonstrating its involvement in epidermal development18.
To evaluate how well the roles of the identified genes are understood in epidermal development, we queried the PubMed literature database and examined the results. For each gene, the keyword used in the PubMed search was constructed as “<symbol>[tiab] AND (epidermis OR skin)”. The search results showed that a large proportion of identified genes (\( \sim 42 \% \) = 34/81) have no publications related to skin. Therefore, these understudied novel genes revealed potential candidate genes for new studies on epidermal development. In addition, the majority (\( > 70 \% \)) of identified genes were not in the epidermis development GO term (Fig. S2). These novel genes demonstrate the ability of the paradigm to discover unknown factors in epidermal development.
To demonstrate the effectiveness of the paradigm computationally, top-ranked genes using collective comparisons were compared to genes using individual comparisons (Text S1). Fig. S3 shows the significantly (p-value = \(3.6\,\times \,{10}^{-9}\)) increased epidermal development genes identified by the paradigm compared to differentially expressed genes derived from individual comparisons.
Validation of Sbsn role in epidermal differentiation by loss-of-function and other experiments
Among the identified genes, a top gene (SBSN) (with a high consensus score of 9) was selected to validate its role in epidermal development. A phylogenetics-based GO analysis revealed enriched GO terms related to epidermal development using co-evolved genes of SBSN (Text S2, Fig. S4). In addition, a time-course microarray dataset showed an increased expression of SBSN upon epidermal differentiation (Text S3, Fig. S5). These results suggest a potentially critical role of SBSN in epidermal development. To determine the cellular component that Sbsn is involved with, we performed a study of the differentially expressed genes in differentiating mouse primary keratinocyte cultures from mice with Sbsn knockdown. In Sbsn knockdown mouse cultures, 326 genes were up-regulated, and 161 genes were down-regulated (Methods, Table S3, Fig. 4a). To investigate the functional roles of Sbsn, these differentially expressed genes were used to search for enriched GO terms19 using Fisher’s exact test (null hypothesized log-odds-ratio \( < 2\)) with the genes expressed in the Sbsn knockdown mouse culture and the controls as background. Specifically, the cornified envelope GO term was found enriched in the genes down-regulated upon Sbsn knockdown (p-value < 0.05), and eight cornified envelope genes were down-regulated (Table S4). These results suggest the role that Sbsn may play in epidermal differentiation and cornified envelope formation.

Validations of SBSN in epidermal differentiation. (a) Heatmap of the expression levels between Sbsn knockdown mice and controls. Expression levels are shown for genes differentially expressed (under |log2-fold-change| >0.5 and q-value < 0.05) upon Sbsn knockdown. Red and white colors indicate high and low expression levels (arc-sine hyperbolic transformed normalized counts by DESeq and scaled by standard deviations) for 326 up-regulated genes, respectively. Blue and white colors indicate high and low expression levels for 161 down-regulated genes, respectively. (b) Expression values of SBSN normalized by 18 S rRNA in differentiated keratinocytes upon IL-4 treatment. To evaluate the gene expression changes of SBSN during keratinocyte differentiation upon IL-4 treatment, an RT-PCR experiment was performed with nine differentiated cells with and without IL-4 treatments (three replicates per condition). The expression values of SBSN were normalized by the expression levels of 18 S rRNA. The boxplot shows a significant decrease of SBSN expression at two IL doses (5 ng/ml and 50 ng/ml) (*p-value \( < 0.05\)). (c) Expression values of full-length SBSN transcript (v1) in AD skins. To evaluate the expression changes of SBSN in AD skins, expression values were measured in AD skins for the SBSN transcripts via RT-PCR. The expression levels were normalized by the expression levels of G6PD. The full-length SBSN transcript showed significantly decreased expression levels in AD lesional skins compared to AD nonlesional and control skins (***p-value \( < 0.001\), **p-value \( < 0.01\), *p-value \( < 0.05\)).
Atopic dermatitis (AD) is the most common chronic inflammatory skin disease20. IL-4, a type 2 cytokine, contributes to the development of AD. Because broad defects of cornified envelope have been identified in AD21, SBSN may play a critical role in AD via defective cornification. To investigate the putative role of SBSN in AD, differentiated primary normal human epidermal keratinocytes (NHEKs) were cultured to examine the expression levels of SBSN upon IL-4 treatments via RT-PCR. In the presence of IL-4 (at doses of 5 ng/ml and 50 ng/ml), SBSN mRNA levels in the differentiated cells were significantly decreased as compared to differentiated cells without cytokine treatment (Fig. 4b). These decreased expression levels of SBSN upon IL-4 treatment suggest a critical precursor role of SBSN in the development of AD via disruption of cornification—and further indicate an important role of SBSN in epidermal differentiation.
To investigate the role of SBSN in AD, expression levels of three SBSN transcripts were measured in AD lesional/nonlesional and control skins via RT-PCR (Methods). A total of 49 skin biopsies were measured, consisting of 16 AD lesional skin biopsies, 16 AD nonlesional skin biopsies, and 17 healthy controls. The expression levels of SBSN transcripts were normalized to G6PD. SBSN transcript v1 (NM_001166034.1) showed a significantly decreased level in AD lesional skin compared to AD nonlesional skin and controls (Fig. 4c). The decreased expression levels of the full-length transcript of SBSN suggests an important role of this SBSN isoform in AD.
Generalization of the paradigm as demonstrated by its application on CIT
To investigate the generalizability of our integrative analysis approach, we applied the paradigm to reveal thermogenesis genes in tissues upon cold exposure. We collected ten gene expression datasets from GEO (Table S5). These gene expression data were collected from tissues of mice treated with cold temperature to induce thermogenesis. Both microarray and RNA-Seq data were collected. Because thermogenesis is always activated upon cold exposure, the direction of thermogenesis is thus increased in all the 24 comparisons within the ten collected datasets. Using DEG analysis, the paradigm calculated the consensus scores for measured genes from 24 comparisons and identified 153 genes with a consensus score \(\ge 6\) (Table S6). These 153 identified genes were then used to perform GO analysis. Enriched GO terms are related to energy homeostasis (Fig. S6). Literature curation confirmed the functional evidence in CIT of some identified genes. For example, elongation of very-long-chain fatty acids (Elovl3, consensus score = 13) in ablated mice showed a proliferated metabolic rate in a cold environment, indicating a higher capacity for brown fat-mediated nonshivering thermogenesis. Thus, Elovl3 is a key regulator for CIT in adipose tissue upon cold exposure22. As another example, carnitine palmitoyltransferase 2 (Cpt2, consensus score = 11) depletion mediates the fatty acid oxidation in adipose tissue, which is required for CITs, suggesting the critical role of Cpt2 in CIT15,23. This second application of our paradigm in CIT suggests that the paradigm can be generalized to other biological processes. Our paradigm is a simple but important integrative data processing approach for gene expression data.
Discussion
We propose a gene expression data processing paradigm to identify key factors in biological processes. The collection of gene expression data enhances the identification of key factors in biological processes. The application of the paradigm for epidermal development revealed known and novel epidermal development genes. To validate the novel predictions, an understudied gene, SBSN, was specifically investigated for its potential role in epidermal differentiation. SBSN has been identified in the suprabasal layers of the epithelia in the epidermis24. Although SBSN was previously shown to be induced upon differentiation of epidermal keratinocytes, no loss-of-function study has been performed to demonstrate the functional role of SBSN in epidermis or skin. Our phylogenetics-based GO analysis suggests relevant biological processes in epidermal development for SBSN co-evolved genes (Fig. S4). RNA-Seq analysis in Sbsn knockdown mouse keratinocyte cultures revealed down-regulated cornified envelope genes, suggesting a role for Sbsn in epidermal differentiation. SBSN may also be critical in AD, an inflammatory skin disease, because AD has a broadly defective cornified envelope21,25. Due to the full-length isoform of SBSN potentially playing a more critical role in the development of AD (Text S4, Fig. 4c and S7) and IL-4 being involved in AD20, we examined the effects of IL-4 on human differentiating keratinocyte cultures and found decreased expression levels of SBSN in IL-4 treated compared to non-treated cultures (Fig. 4b). These results indicate that SBSN may be a target for aberrant cytokine production in AD.
The paradigm identified key genes by collecting multiple datasets and integrating information from the collected datasets, especially from datasets of the cell or tissue types most relevant to the target biological process. However, due to the limited availability of such datasets in certain areas, relevant datasets from other cell or tissue types may also be used, for they generally will not worsen the results. This is consistent with the idea of ensemble learning, in which merging many weak and independent classifiers will result in a strong classifier26. The heatmap of identified genes (consensus score \(\ge 6\)) showed seven experimental comparisons from epidermal cells clustered together (Fig. S1). To systematically cluster the 24 experimental comparisons, a hierarchical clustering analysis using an affinity distance metric (Text S5) grouped the comparisons into eleven distinct clusters (at a cutoff distance \(\le 0.05\)) (Fig. S8). As a result, the seven experimental comparisons from epidermal cells were also consistently clustered together in the hierarchical clustering analysis. These results indicate that the experimental comparisons from epidermal cells contributed the most. In the future, with more datasets from epidermal tissues/cells generated, it may not be necessary to include datasets from nonepidermal tissues/cells, as the marginal contribution from them is likely to be negligible.
The paradigm starts from gene expression datasets with the perturbation of a biological process. This data collection process is critical. As for the application in epidermal development, we searched ArrayExpress using the text of epidermal development for candidate gene expression datasets. However, none of the five retrieved datasets showed changes in epidermal development, leaving us with no data from reliance on the bare keyword search functionality offered by ArrayExpress (Fig. S9). Because known genes annotated in the epidermis development GO term provide candidate factors responsible for the regulation of epidermal development, datasets with these perturbed known genes can be a starting point for our paradigm. However, it should be understood that the paradigm is not limited to a single GO term. Due to incomplete annotation in GO5, genes in other GO terms, such as keratinocyte differentiation (GO:0030216) and epidermal cell differentiation (GO:0009913), can also play roles in epidermis development. Thus, starting from these additional genes along with the genes in the epidermis development GO term, the performance of the paradigm is expected to improve because more information may be borrowed from other relevant datasets. To apply our paradigm, it is critical to examine the collection of gene expression datasets. In addition, our paradigm can include both microarray and RNA-Seq data, as shown in the CIT application, enabling the inclusion of more data—leading to better results than with only one data type.
To obtain a manageable number of identified genes, our computation analysis focused on genes with a consensus score \(\ge 6\). A simulation was performed to evaluate the empirical distribution of consensus scores in epidermal development and CIT, and a cutoff of \(\ge 6\) corresponds to an empirical p-value = \(3.72\,\times \,{10}^{-8}\) and \(1.09\,\times \,{10}^{-8}\) for epidermal development and CIT, respectively (Text S6 and Fig. S10). But other thresholds may also be used. Higher thresholds lead to fewer but more robust identified genes, while lower thresholds lead to more but less robust identified genes. An investigator should pick a cutoff appropriate for the intent of the investigation. For example, if the purpose is to identify more novel epidermis development genes for further experimental validation, genes with consensus scores lower than 6 can also be considered. In addition, the cutoff should be also related to the total number of comparisons used in the analysis. In general, with more comparisons, the cutoff for the consensus score should be greater.
It is worth mentioning that genes with negative consensus scores, in general, do not contribute to regulation in biological processes. For the application of epidermal development and CIT, a consensus score cutoff ≤−5 is used to extract negative genes, and no enriched GO terms are related to epidermal development (44 negative genes) and CIT (95 negative genes) (data not shown). These results are consistent with the intent of the scoring scheme defined in Fig. 1. The number of genes with positive and negative consensus scores would be expected to be roughly the same due to normalization, but the positive genes, in fact, have longer tails than the negative genes (Fig. S11). Quantile testing shows significantly larger positive scores compared to the absolute negative score at 0.95-quantile for both applications (p-value < \(2.2\,\times \,{10}^{-16}\))27. This suggests that genes positively correlated with epidermal development and CIT are more likely to be consistent across different experiments, indicating that these positive genes are more likely to be relevant to the respective processes. In summary, the paradigm is valuable in identifying key factors for biological processes using gene expression data.
Methods
Ethics statement
All skin samples were collected according to procedures (NKEBN/486/2011) previously approved by the local ethics committee (Independent Bioethics Commission for Research at Medical University of Gdansk). Written consent was obtained from all patients prior to enrollment in the study.
Curating gene expression data related to epidermal development
We collected gene expression datasets related to epidermal development by manual curation according to the following procedure. First, we searched ArrayExpress using the keyword (“epidermis + development” OR “epidermal + development”) AND organism: “homo sapiens”, retrieving only five studies, none of which could be reused to study the epidermal development process because of no change in epidermal development in the datasets (Fig. S9). Therefore, we started from known epidermal development genes to curate datasets with the process perturbed. Specifically, genes from the GO19 epidermis development (accession GO:0008544) term were extracted first for humans. Then, the official symbol of each gene was queried on ArrayExpress for human microarray datasets. Each retrieved dataset was manually examined to retain only the datasets with at least one epidermis development gene being perturbed (i.e., knocked out, knocked down, or overexpressed). To ensure proper downstream statistical analysis, any dataset with no replicates was discarded.
Data processing paradigm of the perturbed expression data
To identify the genes related to a biological process, our data processing paradigm was performed on the gene expression data to capture the affinities between specific genes and the biological process. An affinity score of \(+1\) or \(-1\) means that the gene is positively or negatively related to the biological process. Specifically, if the expression of a gene is increased or decreased in a biological process that is increased, the gene has an affinity score of \(+1\) or \(-1\) for the biological process. Alternatively, if the biological process is decreased, these genes have an affinity score of \(-1\) or \(+1\). The affinity score was 0 or NA for the genes not differentially expressed or unmeasured. The detailed workflow of the paradigm is shown in Fig. 1. For a biological process, systematic data curation is performed to collect gene expression datasets with the process perturbed (increased or decreased). Using DEG analysis (Text S7 and S8)28,29,30, affinity scores are calculated for each gene in each comparison in each dataset. Finally, a consensus score is calculated by summing these affinity scores among the comparisons for each gene. High consensus scores suggest that the corresponding genes are potentially critical to the biological process. Thus, our paradigm is a general framework that can be used to identify the key factors in a biological process.
NHEK cell culture and treatment
Primary NHEKs of neonatal foreskin were purchased from Thermo Fisher Scientific and were maintained in EpiLife Medium containing 0.06 mM CaCl2 and S7 supplemental reagent under standard tissue culture conditions. The cells were seeded in 24 well dishes at 2 × 105/well to form a confluent monolayer. In the following day, the cells were subjected to differentiation by increasing CaCl2 to 1.3 mM in the culture media with or without the human recombinant IL-4 at designated concentrations. The cells were harvested for total RNA extraction before differentiation and were differentiated for 5 days. Total RNA was extracted using RNeasy mini kit according to manufacturer guidelines (QIAGEN, MD). RNA was then reverse transcribed into cDNA using superScript® III reverse transcriptase from Invitrogen (Portland, OR) and was analyzed by real-time RT-PCR using an ABI Prism 7000 sequence detector (Applied Biosystems, Foster City, CA). Primers and probes for human SBSN (Hs01078781_m1) and 18 s (Hs99999901_S1) were purchased from Applied Biosystems (Foster City, CA). Quantities of all target genes in test samples were normalized to the corresponding 18 S levels.
Sbsn siRNA in mouse keratinocyte culture and RNA sequencing
Primary murine keratinocytes were isolated from BALB/c neonatal mice as previously described31. Primary murine keratinocytes were cultured in a supplemented minimal essential medium (Gibco, Thermo Fisher Scientific Inc., Waltham, MA, USA) with 8% fetal bovine serum (FBS, Atlanta Biologicals, Flowery Branch, GA, USA) and 1% antibiotic (Penicillin Streptomycin Amphotericin B, Sigma), with 0.05 mM Ca2+ concentration. A total of \(1\,\times \,{10}^{6}\) cells were seeded in each well of six well plates. Twenty-four hours after seeding, the siRNA mix (Opti-Mem serum free media (GIBCO), 75 pmol of siRNA (Dharmacon), and HiPerfect transfection reagent (Qiagen)) was added to cells. For SBSN, the siRNA used were Dharmacon, SMARTpool; siGENOME Sbsn siRNA M-054578-01-0005 and Control (mouse) SMARTpool; and siGENOME Non-Targeting siRNA Pool #1. Each condition was done in triplicates. To induce keratinocyte differentiation, a final concentration of 0.12 mM Ca2+ was used. RNA was harvested 48 hours after siRNA transfection. Total RNA from cells was extracted using RNeasy kit (Qiagen) according to manufacturer instructions. A total of 100 ng was used to prepare the libraries utilizing a Neoprep Library kit (Illumina). RNA sequencing was performed in the NIAMS Genome Core Facility at the National Institutes of Health.
DEG analysis using Sbsn knockdown RNA-Seq data in mouse differentiating primary keratinocyte cultures
To identify the differentially expressed genes in mouse differentiating primary keratinocyte cultures in which Sbsn had been knocked down with siRNA, the following analysis was performed. The raw RNA-Seq reads were aligned to the mouse (mm10) genome using STAR (version 2.5.1b)32 with default settings. The uniquely aligned reads were retained to calculate the read counts for each gene against the UCSC KnownGene annotation (mm10), and a count table was constructed by counting the number of reads aligned uniquely to each of the genes for each sample. DEG analysis was performed by DESeq233. To adjust the batch effect, a generalized linear model with a batch factor was used to model the read counts for all samples, and the Wald test was used to test the significance of differences in gene expression between Sbsn knockdown samples and controls. FDR adjusted q-values were then calculated from the p-values in the Wald test using the Benjamini-Hochberg procedure34. The log2-fold changes between Sbsn knockdown samples and controls were also calculated for each gene. The differentially expressed genes were identified under |log2-fold-change| \( > 0.5\) and \(q < 0.05\).
RT-PCR analysis of AD skin
For the current study, arm skin samples (2 mm punch biopsies of 3 mm depth) were taken from AD patients (from lesional and nonlesional AD skin), and skin samples (controls) were obtained from healthy subjects. The nonlesional skin biopsy was performed at a 10 cm distance (at least) from AD skin lesions. Immediately after biopsy, the skin samples were placed in RNA-later solution (Qiagen) and were stored at –20 0C. Total RNA was isolated using standard methods. The mRNA levels were analyzed by real-time RT-PCR with TaqMan primer-probe sets using the Path-ID Multiplex One-Step RT-PCR kit (Path-ID™ Multiplex One-Step RT-PCR Kit (Applied Biosystems). The reference transcript G6PD was used as an internal standard and was amplified together with each target gene transcript in the same well using primers and probes, as shown in Table S7. The level of each analyzed transcript was normalized to that of the appropriate reference transcript.
Data availability
The datasets used in this study are available in the National Center for Biotechnology Information’s (NCBI’s) GEO2 with accession GSE100100.
References
Rustici, G. et al. ArrayExpress update–trends in database growth and links to data analysis tools. Nucleic Acids Res 41, D987–990, https://doi.org/10.1093/nar/gks1174 (2013).
Edgar, R., Domrachev, M. & Lash, A. E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30, 207–210 (2002).
Qin, B. et al. CistromeMap: a knowledgebase and web server for ChIP-Seq and DNase-Seq studies in mouse and human. Bioinformatics 28, 1411–1412, https://doi.org/10.1093/bioinformatics/bts157 (2012).
Guo, Z. et al. RNASeqMetaDB: a database and web server for navigating metadata of publicly available mouse RNA-Seq datasets. Bioinformatics 31, 4038–4040, https://doi.org/10.1093/bioinformatics/btv503 (2015).
Li, J. et al. SFMetaDB: a comprehensive annotation of mouse RNA splicing factor RNA-Seq datasets. Database 2017, bax071–bax071, https://doi.org/10.1093/database/bax071 (2017).
Li, J. et al. RBPMetaDB: A comprehensive annotation of mouse RNA-Seq datasets with perturbations of RNA-binding proteins. Database (Oxford), https://doi.org/10.1093/database/bay054 (in press).
Li, Z., Li, J. & Yu, P. GEOMetaCuration: a web-based application for accurate manual curation of Gene Expression Omnibus metadata. Database 2018, bay019–bay019, https://doi.org/10.1093/database/bay019 (2018).
Bhaduri, A. et al. Network Analysis Identifies Mitochondrial Regulation of Epidermal Differentiation by MPZL3 and FDXR. Dev Cell 35, 444–457, https://doi.org/10.1016/j.devcel.2015.10.023 (2015).
Churchill, G. A. Fundamentals of experimental design for cDNA microarrays. Nature genetics 32, 490–495 (2002).
Kavvoura, F. K. & Ioannidis, J. P. Methods for meta-analysis in genetic association studies: a review of their potential and pitfalls. Human genetics 123, 1–14 (2008).
Elias, P. M. The skin barrier as an innate immune element. Semin Immunopathol 29, 3–14, https://doi.org/10.1007/s00281-007-0060-9 (2007).
Li, J. & Yu, P. Genome-wide transcriptome analysis identifies alternative splicing regulatory network and key splicing factors in mouse and human psoriasis. Scientific Reports 8(1) (2018).
Thomas, P. D. The Gene Ontology and the Meaning of Biological Function. Methods Mol Biol 1446, 15–24, https://doi.org/10.1007/978-1-4939-3743-1_2 (2017).
Ellis, J. M. et al. Adipose acyl-CoA synthetase-1 directs fatty acids toward beta-oxidation and is required for cold thermogenesis. Cell metabolism 12, 53–64, https://doi.org/10.1016/j.cmet.2010.05.012 (2010).
Li, J., Deng, S.-P., Wei, G. & Yu, P. CITGeneDB: a comprehensive database of human and mouse genes enhancing or suppressing cold-induced thermogenesis validated by perturbation experiments in mice. Database 2018, bay012–bay012, https://doi.org/10.1093/database/bay012 (2018).
Hyter, S. et al. Endothelin-1 is a transcriptional target of p53 in epidermal keratinocytes and regulates ultraviolet-induced melanocyte homeostasis. Pigment Cell Melanoma Res. 26, 247–258, https://doi.org/10.1111/pcmr.12063 (2013).
Vasireddy, V. et al. Loss of functional ELOVL4 depletes very long-chain fatty acids (≥C28) and the unique ω-O-acylceramides in skin leading to neonatal death. Human molecular genetics 16, 471–482 (2007).
Yang, J. M., Sim, S. M., Kim, H. Y. & Park, G. T. Expression of the homeobox gene, HOPX, is modulated by cell differentiation in human keratinocytes and is involved in the expression of differentiation markers. Eur J Cell Biol 89, 537–546, https://doi.org/10.1016/j.ejcb.2010.01.005 (2010).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25, 25–29, https://doi.org/10.1038/75556 (2000).
Brunner, P. M., Guttman-Yassky, E. & Leung, D. Y. The immunology of atopic dermatitis and its reversibility with broad-spectrum and targeted therapies. J Allergy Clin Immunol 139, S65–S76, https://doi.org/10.1016/j.jaci.2017.01.011 (2017).
Guttman-Yassky, E. et al. Broad defects in epidermal cornification in atopic dermatitis identified through genomic analysis. J Allergy Clin Immunol 124, 1235–1244 e1258, https://doi.org/10.1016/j.jaci.2009.09.031 (2009).
Westerberg, R. et al. ELOVL3 is an important component for early onset of lipid recruitment in brown adipose tissue. The Journal of biological chemistry 281, 4958–4968, https://doi.org/10.1074/jbc.M511588200 (2006).
Lee, J., Ellis, J. M. & Wolfgang, M. J. Adipose Fatty Acid Oxidation Is Required for Thermogenesis and Potentiates Oxidative Stress-Induced Inflammation. Cell reports 10, 266–279, https://doi.org/10.1016/j.celrep.2014.12.023 (2015).
Park, G. T., Lim, S. E., Jang, S.-I. & Morasso, M. I. Suprabasin, a novel epidermal differentiation marker and potential cornified envelope precursor. Journal of Biological Chemistry 277, 45195–45202 (2002).
Trzeciak, M. et al. Expression of Cornified Envelope Proteins in Skin and Its Relationship with Atopic Dermatitis Phenotype. Acta Derm Venereol 97, 36–41, https://doi.org/10.2340/00015555-2482 (2017).
Hastie, T., Tibshirani, R. & Friedman, J. The Elements of Statistical Learning: Data Mining, Inference, and Prediction. (Springer, 2009).
Conover, W. J. Practical nonparametric statistics. 3rd ed. 396–406 (Wiley. pp. 396–406, 1999).
Qian, X. et al. RNA-seq analysis of glycosylation related gene expression in STZ-induced diabetic rat kidney inner medulla. Frontiers in Physiology 6 (2015).
Guo, Z. et al. Possible mechanisms of host resistance to Haemonchus contortus infection in sheep breeds native to the Canary Islands. Scientific Reports 6(1) (2016).
Osenberg, S. et al. Activity-dependent aberrations in gene expression and alternative splicing in a mouse model of Rett syndrome. Proc Natl Acad Sci USA, https://doi.org/10.1073/pnas.1722546115 (2018).
Palazzo, E. et al. The homeoprotein DLX3 and tumor suppressor p53 co-regulate cell cycle progression and squamous tumor growth. Oncogene 35, 3114–3124, https://doi.org/10.1038/onc.2015.380 (2016).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21, https://doi.org/10.1093/bioinformatics/bts635 (2013).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq. 2. Genome Biol 15, 550, https://doi.org/10.1186/s13059-014-0550-8 (2014).
Benjamini, Y. & Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society. Series B (Methodological) 57, 289–300 (1995).
Acknowledgements
The authors thank Yu Chen (ECE Department at Texas A&M University), Veronica Nagle (Laboratory of Skin Biology, NIAMS, NIH), and Stephen Brooks from the NIAMS Biodata Mining core facility (NIAMS, NIH) for their contributions to this work. This work was supported by startup funding to PY from the ECE department and Texas A&M Engineering Experiment Station/Dwight Look College of Engineering at Texas A&M University, by funding from TEES-AgriLife Center for Bioinformatics and Genomic Systems Engineering (CBGSE) at Texas A&M University, and by TEES seed grant. DYL was supported by NIH grants R01 AR41256 and U19 AI117673. This work was supported by IRP NIH ZIA-AR041124 (MIM), R01AR051930 (TM) and R01AG028492 (PE), which were administered by the Northern California Institute for Research and Education and with resources of the Research Service, Department of Veterans Affairs. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
J.L., L.Z. and P.Y. carried out the analyses and wrote the manuscript. A.U., L.B., T.M., P.E., T.P., M.SB., M.T., D.L. and M.M. performed validation experiments. All the authors reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, J., Zheng, L., Uchiyama, A. et al. A data mining paradigm for identifying key factors in biological processes using gene expression data. Sci Rep 8, 9083 (2018). https://doi.org/10.1038/s41598-018-27258-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-27258-8
This article is cited by
-
Identification and validation of core genes as promising diagnostic signature in hepatocellular carcinoma based on integrated bioinformatics approach
Scientific Reports (2022)
-
Suprabasin: Role in human cancers and other diseases
Molecular Biology Reports (2022)
-
Identification of hub genes associated with obesity-induced hepatocellular carcinoma risk based on integrated bioinformatics analysis
Medical Oncology (2021)
-
Suprabasin-null mice retain skin barrier function and show high contact hypersensitivity to nickel upon oral nickel loading
Scientific Reports (2020)
-
Integrated analysis of a compendium of RNA-Seq datasets for splicing factors
Scientific Data (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
