
Abstract
We re-evaluate the Raman spectroscopic quantification of the molar ratio and pressure for CH4–CO2 mixtures. Firstly, the Raman quantification factors of CH4 and CO2 increase with rising pressure at room temperature, indicating that Raman quantification of CH4/CO2 molar ratio can be applied to those fluid inclusions (FIs) with high internal pressure (i.e., > 15 MPa). Secondly, the v1(CH4) peak position shifts to lower wavenumber with increasing pressure at constant temperature, confirming that the v1(CH4) peak position can be used to calculate the fluid pressure. However, this method should be carefully calibrated before applying to FI analyses because large discrepancies exist among the reported v1(CH4)-P curves, especially in the high-pressure range. These calibrations are applied to CH4-rich FIs in quartz veins of the Silurian Longmaxi black shales in southern Sichuan Basin. The vapor phases of these FIs are mainly composed of CH4 and minor CO2, with CO2 molar fractions from 4.4% to 7.4%. The pressure of single-phase gas FI ranges from 103.65 to 128.35 MPa at room temperature, which is higher than previously reported. Thermodynamic calculations supported the presence of extremely high-pressure CH4-saturated fluid (218.03–256.82 MPa at 200 °C), which may be responsible for the expulsion of CH4 to adjacent reservoirs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Fluid inclusions (FIs) recorded the chemical compositions of paleo-geofluids and could provide important information for the relevant physicochemical conditions (e.g., temperature, pressure, pH, Eh) of geological processes (e.g., Roedder and Bodnar 1980; Bergman and Dubessy 1984; Roedder 1984; Petrychenko et al. 2005). In the field of petroleum geology, in combination with the construction of burial history, microthermometric analyses of FIs hosted in authigenic reservoir cements have been applied to investigate the hydrocarbon accumulation process, including the timing and the temperature–pressure (T–P) conditions (e.g., Burruss et al. 1983; Bodnar 1990; Thiéry et al. 2000; Munz 2001; Cao et al. 2006; Guo et al. 2012; Cui et al. 2013; Li et al. 2015; Chen et al. 2016; Fang et al. 2017). Methane (CH4) and carbon dioxide (CO2) are common components in the vapor phase of hydrocarbon-bearing FIs trapped under elevated T–P conditions (e.g., Pironon et al. 2001; Dutkiewicz et al. 2003; Lüders et al. 2012). Then trying to fathom the composition of the CH4–CO2 mixture is required for the measurements of the fluid composition, which is also essential for revealing fluid-trapping T–P conditions.
Conventionally, the composition of a FI can be obtained through microthermometric measurements (e.g., Collins 1979; Roedder 1984; Darling 1991; Bodnar 1993). However, this method is difficult to be applied to small FIs with diameter of < 5 µm, and the complex composition of geofluids cannot be accurately measured based on phase diagrams of simple fluid systems (Hollister and Crawford 1981; Mernagh and Wilde 1989; Chou et al. 1990; Walter et al. 2017). Alternatively, in situ Raman spectroscopy can provide quantitative information of FIs based on systemic experimental calibrations (e.g., Pasteris et al. 1988; Mernagh and Wilde 1989; Chou et al. 1990; Lu et al. 2007; Wang et al. 2011). This method is fast and nondestructive, and the spatial resolution of a microscopic confocal Raman spectrometer can reach 2–3 µm.
The intensity of a Raman active species, i, is a function of many factors (see Schmidt 2009). Wopenka and Pasteris (1986) simplified the expression of the Raman intensity of species i as:
where A is the integrated Raman intensity (i.e., peak area), C is the number of Raman active molecules in the analyzed volume, η is the instrument efficiency factor, σ is the Raman scattering coefficient, and IL is the intensity of incident light. For two Raman active components, a and b, in a homogeneous gas phase, their molar ratios can be calculated based on their Raman peak area ratios (Wopenka and Pasteris 1987; Pasteris et al. 1988; Chou et al. 1990):
where Fa/Fb is the Raman quantification factor of a relative to b. In general, the reported Raman quantification factors of gaseous i are relative to N2. For example, FCH4 is the Raman quantification factor of CH4 relative to N2. Theoretically, if the Raman quantification factors of CH4 and CO2 are known, we can measure the molar ratios between CH4 and CO2 in CH4–CO2 mixtures through Raman spectroscopic analyses. However, the reported FCH4 and FCO2 are quite different among different laboratories (Wopenka and Pasteris 1987; Seitz et al. 1993, 1996; Xi et al. 2014), and the effect of fluid pressure still needs further investigation (c.f., Chou et al. 1990; Seitz et al. 1993, 1996). In pure CH4 system, the symmetric stretching vibration band of CH4 (v1) shifts to lower wavenumber with rising pressure at constant temperature (Lin et al. 2007; Lu et al. 2007; Shang et al. 2015). Therefore, the wavenumber of v1(CH4) band can be used to calculate the pressure of CH4. In fact, the v1(CH4)-P curve can also be applied to CH4-dominated dry gas. For example, the experimental works of Seitz et al. (1993, 1996) demonstrated that the presence of minor amount of CO2 or N2 does not change the v1(CH4)-P curve significantly. Because the reported v1(CH4)-P curves are quite different, Lu et al. (2007) constructed a “unified” v1(CH4)-P curve for calculation of the CH4 pressure. This method needs an accurate determination of the v1(CH4) wavenumber of low-pressure CH4 (e.g., < 1 bar). Considering the experimental conditions which are different among different laboratories, a more practical method is to construct the laboratory-based v1(CH4)-P calibration curve.
In this study, we used fused silica capillary tubing as spectroscopic cell (Chou et al. 2005; Wang et al. 2011). CH4 and CH4–CO2–CO–N2 mixtures with known composition were loaded into the silica tubing and pressurized to pressures up to 136 MPa. In situ Raman spectra of these gases were collected at room temperature (22 °C). The Raman quantification factors, FCH4 and FCO2, were determined, and the v1(CH4)-P curve was constructed. Then, we analyzed the gas composition and pressure of two-phase aqueous and single-phase gas FIs hosted in quartz veins in the Lower Silurian Longmaxi Formation shale in Sichuan Basin. Compared with the available burial history, a paleo-overpressure of CH4-saturated fluid was proposed.
2 Experimental methods
2.1 High-pressure optical cell
The procedures for preparing a high-pressure optical cell (HPOC) were presented in Chou et al. (2005) and Wang et al. (2011). Pure CH4 (99.99% purity) and CH4–CO2–CO–N2 mixture with molar ratio of 1:1:1:2 were purchased from Jiahe Air Chemical (Shanghai) Co. Ltd. CH4 or CH4–CO2–CO–N2 gas was loaded into the cell and then was sealed using mercury at room temperature (Fig. 1; Wan et al. 2017). In this study, water was used as pressure medium. The pressure inside the line and cell was adjusted by a pressure generator. Pressures were measured by an Omega PX91N0-35KSV digital pressure transducer with Omega DP41-S-230 manometer (241 MPa full scale, accurate to ± 0.5%). The experimental setup is shown in Fig. 1. In order to maintain the sample temperature at 22 °C, the HPOC was inserted into a Linkam CAP500 heating–cooling stage (Wan et al. 2015).

A schematic diagram showing the experimental setup
2.2 Raman spectroscopic analyses and spectral processing
In situ Raman spectra of pure CH4 and CH4–CO2–CO–N2 mixture were collected using a high-resolution JY/Horiba Raman spectrometer (LabRaman HR 800). The wavelength of the air-cooled frequency-doubled Nd:YAG excitation laser is 532 nm. A 50 × Olympus long-working distance objective and an 1800 groove/mm grating with spectral resolution of approximately 1 cm−1 were applied. The spectrometer was calibrated with the 520.7 cm−1 peak of silicon. For the pure CH4 system, the Ne emission lines at 2835.22 and 3006.36 cm−1 were also collected simultaneously (Fig. 2). Then, the v1(CH4) peak position was calibrated following the procedures presented in Kim et al. (1986) and Fang et al. (2018).

Typical spectra of pure CH4 (upper) and CH4–CO2–CO–N2 mixture (lower). Note that the neon emission lines at 2835.22 and 3006.36 cm−1 are collected simultaneously to calibrate the v1(CH4) peak position for pure CH4. For the CH4–CO2–CO–N2 mixture, the spectra of diamond at ~ 1331.70 cm−1 (star) are also collected to calibrate the spectrometer. Gray areas in the lower spectrum showing the ranges of wavenumber for the calculation of the relevant peak areas
For the CH4–CO2–CO–N2 system, the peak areas of v1(CH4) band (ACH4), the upper band of v1–2v2(CO2) (ACO2), and v1(N2) band (AN2) were integrated to calculate FCH4 and FCO2 based on Eq. (2). Because the intensity of the upper band of v1–2v2(CO2) is higher than that of the lower band (Fig. 2), we calculated the peak area of the upper band to obtain FCO2; the uncertainty in peak area calculation can be reduced in this way. The wavenumber ranges for the calculations of ACH4, ACO2, and AN2 are from 2880 to 2950 cm−1, from 1375 to 1400 cm−1, and from 2310 to 2345 cm−1, respectively (Fig. 2).
3 Results
3.1 CH4–CO2–CO–N2 system
As shown in Fig. 3a, the characteristic band positions of all the investigated gases shift slightly to lower wavenumber with increasing pressure. For FCH4 and FCO2, they increase with rising pressure in the low-pressure range (e.g., < 15 MPa; Fig. 3b). For example, FCH4 increases from 5.22 at 1 MPa to 6.37 at 15 MPa; FCO2 increases from 1.46 at 1 MPa to 1.70 at 10 MPa (Table 1). At higher pressures, neither FCH4 nor FCO2 shows significant variations.

a Typical Raman spectra of CH4–CO2–CO–N2 mixtures collected at pressures between 1 and 60 MPa and at constant temperature of 22 °C and b the obtained Raman quantification factors of CH4 and CO2 as a function of pressure at 22 °C. Data plotted are those listed in Table 1. Previous works of Wopenka and Pasteris (1987), Chou et al. (1990), Seitz et al. (1993, 1996) and Xi et al. (2014) are also shown for comparison
3.2 Pure CH4 system
Figure 4a shows the v1(CH4) spectra collected at pressures from 0.2 to 136 MPa and at constant temperature of 22 °C. The v1(CH4) peak position shifts from 2916.43 cm−1 at 0.2 MPa to 2908.81 cm−1 at 136 MPa (Table 2). Figure 4b further demonstrates the dependence of v1(CH4) peak position on pressure in pure CH4. In this study, the v1(CH4)-P curve can be expressed as:

a Typical Raman spectra of pure CH4 collected at pressures between 0.2 and 136 MPa and at constant temperature of 22 °C. Neon emission lines are also collected to calibrate the v1(CH4) peak position and b the dependence of v1(CH4) peak position on fluid pressure at 22 °C. Data plotted are those listed in Table 2. Previous works of Chou et al. (1990), Seitz et al. (1996), Thieu et al. (2000), Hansen et al. (2001), Jager and Sloan (2001) and Lu et al. (2007) are also shown for comparison
4 Discussion
4.1 Comparisons with previous experimental calibrations
Previously experimental calibrations of FCH4 and FCO2 were also carried out at room temperatures (e.g., Wopenka and Pasteris 1987; Chou et al. 1990; Seitz et al. 1993, 1996). Wopenka and Pasteris (1987) collected the spectra of various gas mixtures with known composition at pressures of < 1.5 MPa. The FCH4 and FCO2 were reported to be 6.7 ± 0.15 and 1.52 ± 0.047, respectively (Wopenka and Pasteris 1987). The experiments of Chou et al. (1990) and Seitz et al. (1993) were carried out in the same laboratory as those for Wopenka and Pasteris (1987). Chou et al. (1990) collected the spectra of equimolar CH4–N2 mixture at pressures up to 65 MPa. They found that FCH4 increases with rising pressure at < ~7 MPa and reaches a relatively stable value of ~ 7.1 above 7 MPa. Seitz et al. (1993) obtained similar FCH4 of 7.39 ± 0.20. It should be noted that they found FCH4 was independent of composition in the CH4–N2 mixtures, given that the pressure is high enough (e.g., > 15 MPa). Our experiments confirmed that FCH4 and FCO2 are dependent on fluid pressure at low pressure, but keep relatively constant at high pressure (Fig. 3b). This phenomenon could be interpreted in the context of the molecular interactions (Seitz et al. 1993). As listed in Eq. (2), the Raman quantification factor is the product of the instrumental efficiency and the molecular parameters. Through monitoring the F-factor (e.g., FCH4), the change in the relative scattering efficiencies of the two components (i.e., CH4 and N2) can be monitored as a function of the molecular environment, since all other parameters are held constant (Seitz et al. 1993). That is to say, the effect of molecular interactions at low pressures is stronger than those at high pressures. Therefore, before applying the F-factor to measure the compositions of natural FIs, the experimental calibrations must be carried out in a broad pressure range that allows the determination of the steady F-factor.
Xi et al. (2014) obtained FCH4 (9.56) and FCO2 (0.86) for another spectrometer; the quantification factors were reported to be independent of pressure and fluid composition at pressures from 5 to 10 MPa. However, the reported FCH4 is more than 30% higher than those reported in Chou et al. (1990) (Fig. 3b). Because the FCO2 of Wopenka and Pasteris (1987) was obtained at low pressures that the molecular effect is significant, the experimental calibrations of Seitz et al. (1996) (FCO2 = 1.43 at > 10 MPa) were chosen for comparison. It can be seen that the FCO2 value of Xi et al. (2014) is ~ 40% lower than that of Seitz et al. (1996) (Fig. 3b). Our results confirmed that the F-factors obtained in different laboratories are different (Fig. 3b). This can be ascribed to the different instrumental efficiencies [c.f., Eq. (2)] for different Raman spectrometers. Therefore, the F-factors are quite laboratory-specific. Carefully experimental calibrations of F-factors should be carried out before applying this method to natural FIs. If a Raman spectrometer is well calibrated, the compositions of the gas mixtures in fused silica capillary capsules (FSCCs) (e.g., Chou et al. 2008; Wang et al. 2011) can be determined. Such FSCCs can be used as reference samples for other laboratories where the Raman spectrometer has not been calibrated yet.
As shown in Fig. 4b, the reported v1(CH4)-P curves are quite different (e.g., Chou et al. 1990; Seitz et al. 1996; Thieu et al. 2000; Hansen et al. 2001; Jager and Sloan 2001; Lu et al. 2007). This discrepancy was ascribed to the use of different reference standards for the calibration of the measured v1(CH4) wavenumber (Lu et al. 2007). However, the reported v1(CH4)-P curves are still different even the same reference standard was chosen (c.f., Thieu et al. 2000; Lin et al. 2007). To eliminate such discrepancy, Lu et al. (2007) proposed to calculate the difference (D) between the wavenumbers of v1(CH4) at elevated pressure (vp) and near-zero pressure (v0).
Then, D was used as the spectral parameter to construct the calibration curve of pressure. Lu et al. (2007) claimed that their D–P curve is a “unified equation” for the calculation of CH4 pressure in the CH4–H2O and CH4 systems. This is true in the low-pressure range (e.g., < 35 MPa), whereas large disagreement exists in the high-pressure range (e.g., > 35 MPa) [see Fig. 4 of Lu et al. (2007)]. In addition, to obtain the accurate vo value, one can just collect the spectra of the low-pressure CH4 gas (e.g., < 0.1 MPa) and determine the v1(CH4) wavenumber through peak fitting. Again, different laboratories may have different vo values due to the selection of different reference standards for the calibration of the Raman spectrometer [see Table 2 of Lu et al. (2007)]. In conclusion, after accurate determination of the laboratory-specific v0, the equation of Lu et al. (2007) can be used to calculate the pressure of the CH4-dominated vapor phase in FIs, if the pressure is below 35 MPa. However, the application of the D–P curve can result in large error, if the pressure of CH4-dominated vapor phase is higher than 35 MPa.
4.2 Application to natural FIs
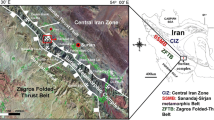
The Lower Silurian Longmaxi Formation is mainly composed of black shales in the Sichuan Basin, southwestern China (Li et al. 2013, 2018; Yang et al. 2019). It is also one of the most important shale gases playing in China at present (Jia et al. 2012; Zou et al. 2015; Jin et al. 2016; Ran et al. 2016). Many studies are focused on the current fluid pressure in the Longmaxi shales (e.g., Zou et al. 2015; Jin et al. 2016; Jiao et al. 2018; Wang et al. 2019), whereas the variation of paleo-fluid pressure has rarely been discussed. Shale gas is different from conventional natural gas in that the formation itself is both the source and the reservoir. Given their source rock nature, the investigation of the paleo-fluid pressure within shales can provide important information on the generation, migration, and accumulation of hydrocarbons. FIs record the T–P conditions of fluid processes in the presence of water and other fluids (e.g., Roedder and Bodnar 1980; Bergman and Dubessy 1984; Roedder 1984). In black shales, FIs can only be observed in some vein minerals, such as calcite and quartz. In this study, the quartz-calcite vein samples of the Lower Silurian Longmaxi shale were collected from an exploratory well (No. X) in southern Sichuan Basin, southwestern China (Fig. 5). The sampling depth is ~ 3830 m. FIs in the quartz veins were observed under a conventional polarizing microscope. Then, typical FIs were chosen for in situ Raman spectroscopic analyses.

A geological map showing the location of Well X [modified after National 1:200,000 Geological Map (H-48-23)]. b The magnification of the black-rectangle-marked area in a
As shown in Fig. 6a, b, two types of FIs, two-phase aqueous inclusions (type I) and single-phase gas inclusions (type II) were identified in the quartz veins. For type I inclusions, the filling degree of the vapor phase ranges from 0.05 to 0.6. The size ranges from 1 to 25 µm. In situ Raman spectroscopic analyses show that the vapor phase is mainly composed of pure CH4 (Fig. 6c; Table 3). The vapor pressure at room temperature can reach 45.25 MPa [c.f., Eq. (3)]. As to type II inclusions, they generally show cogenetic features with type I inclusions. The size ranges from 0.5 to 15 µm. In most cases, the gas inside type II FIs is pure CH4. Only a few contain minor CO2 (4.4–7.4 mol%; Fig. 6d; Table 3). This type of FIs is characterized by extremely high pressure at room temperature; in situ Raman quantification yields CH4 pressure ranging from 103.65 to 128.35 MPa. As discussed in Sect. 4.1, FCH4 and FCO2 are pressure dependent at low pressures. Before applying F-factor to measure the molar ratio of CH4 and CO2, the internal pressure of the FIs should be estimated so that proper F-factor values could be chosen. Because the CH4/CO2 ratio is unknown, it is impractical to get the exact internal pressure (c.f., Chou et al. 1990). Here, we propose a method to estimate the pressure for CH4-dominated gas mixtures at room temperature. Chou et al. (1990) showed that the presence of N2 results in a high-wavenumber shift of the v1(CH4) band for N2–CH4 mixtures at constant total pressure (see Fig. 7). Therefore, the v1(CH4)-P curve for pure CH4 system can provide the minimum total pressure of CH4–N2 mixtures. If this pressure is higher than the threshold pressure for F-factor (i.e., 15 MPa for FCH4 and FCO2 in Fig. 3b), then we can apply the steady F-factors (i.e., FCH4 = 6.42 and FCO2 = 1.69 in this study) to measure the molar ratio of the N2–CH4 mixtures. For CH4–CO2 mixture, Seitz et al. (1996) found that the addition of < 50 mol% CO2 does not change the v1(CH4)-P curve significantly (see Fig. 1a). Therefore, the v1(CH4)-P curve for pure CH4 system can be used to measure the total pressure of CH4-dominated CH4–CO2 mixtures. Based on the above discussion, the molar ratio of CH4/CO2 can be measured using the steady F-factors, and the fluid pressure of CH4–CO2 mixtures can be obtained using Eq. (3) in this study (Table 3).

Microscopic photographs and in situ Raman spectra of typical FIs hosted in quartz veins from Silurian Longmaxi Formation black shales. a, b Two-phase aqueous FIs coexist with single-phase gas FIs; c Typical spectra for two-phase aqueous FIs. The room-temperature pressure of the vapor phase is also shown. v2-H2O and vs-H2O are the bending and stretching vibration modes of water, respectively, and d typical spectra of single-phase gas FIs indicate the presence of CH4–CO2 mixture and pure CH4 within these FIs. Raman quantification of the composition and room-temperature pressure are also shown. Data plotted are those listed in Table 3. V vapor phase, Aq aqueous phase

A diagram showing the burial history of Well X. The gas window and the maximum burial depth are marked using red lines and blue lines, respectively
Coexistence of two-phase aqueous and single-phase CH4 inclusions suggests that these fluid inclusions were trapped in the two-phase immiscible field (Fig. 6a, b; Fall et al. 2012). Under this circumstance, the homogenization temperatures for the two-phase aqueous inclusions represent trapping temperatures (Roedder 1984; Goldstein and Reynolds 1994). Unfortunately, homogenization experiments were not conducted in this study, due to the high internal pressure of these FIs. However, the trapping temperature can still be estimated based on the composition of the hydrocarbon phase and the hydrocarbon generation theory. Firstly, CH4 in the FIs results from primary trapping instead of secondary pyrolysis of previously trapped oil or bitumen. If CH4 is the product of the pyrolysis of liquid hydrocarbons, residual carbon or pyrobitumen should occur on the walls of these FIs (c.f., Xiong et al. 2004). Also, in situ Raman spectroscopic analyses will be hampered by strong fluorescence due to the presence of aromatic molecules (Khorasani 1987; Dutkiewicz et al. 2003). However, these phenomena were not observed in this study. Secondly, the formation of thermogenic dry gas (e.g., > 95 mol% CH4) generally occurs at > 200 °C (Ro > 2%; Tissot and Welte 1984; Hunt 1996). As described earlier, the vapor phase of the investigated FIs is mainly composed of CH4. Although the exact trapping temperature of these CH4-rich FIs is not available at present, a minimum temperature of 200 °C is reasonable. Compared with the burial history of Well X, these FIs should be formed between Early Triassic and Late Cretaceous (Fig. 7).
For type II FIs, the V-x properties can be obtained based on in situ Raman spectroscopic measurements; the composition and pressure at 22 °C are listed in Table 3. For pure CH4 system, the fluid pressure at 200 °C can be calculated on NIST Chemistry WebBook. As to the CH4–CO2 system, the fluid pressure at 200 °C can be obtained based on the thermodynamic models of Bowers and Helgeson (1983) and Bakker (1999). The calculated results are listed in Table 3. It can be seen that the fluid inclusions were trapped in the high temperature (> 200 °C), high pressure (> 218.03–256.82 MPa), and CH4-saturated immiscible fluid.
As shown in Fig. 7, the formation temperature of Lower Silurian reached 200 °C during Early Triassic and increased to its peak temperature of ~ 220 °C during Late Cretaceous. After Late Cretaceous, this area experienced significant uplift, and the current formation temperature is ~ 120–130 °C. The exact timing of fluid trapping is not available due to the lack of the homogenization temperature of type I FIs. However, the maximum burial depth of Lower Silurian is ~ 6450 m, corresponding to a hydrostatic pressure of 64.5 MPa. Therefore, we can still infer that the paleo-pressure coefficient of the Longmaxi Formation can reach > 3.38 between Late Permian and Late Cretaceous. The presence of such paleo-overpressure may result from hydrocarbon generation (Ungerer et al. 1983; Spencer 1987; Liu et al. 2009) and may account for the expulsion of CH4 to adjacent reservoir rocks.
5 Conclusions
Pure CH4 and CH4–CO2–CO–N2 mixture systems were loaded into fused silica tubing and pressurized to 136 MPa at 22 °C. In situ Raman spectra of these gases were collected and processed to address the potential of using Raman spectroscopic analyses to determine the pressure and molar ratio of CH4–CO2 mixtures in natural FIs. The major contributions are:
- 1.
The Raman peak area ratios of CH4 and CO2 can be used to determine the molar ratio of CH4 and CO2 in natural FIs. However, before applying this approach, an estimation of the total pressure is needed. The v1(CH4) peak position can be used to calculate the pressure of CO2–CH4 mixture if the fluid is dominated by CH4. If the pressure is higher than 15 MPa, then an accurate determination of the CO2/CH4 ratio can be obtained.
- 2.
The v1(CH4) peak position shifts to lower wavenumber with increasing pressure at constant temperature. The v1(CH4)-P curve is constructed. Careful calibration should be taken before applying this curve to measure CH4 pressure in natural FIs, because reported v1(CH4)-P curves are different, especially in the high-pressure range.
- 3.
High-pressure CH4-rich FIs are reported in quartz veins from Longmaxi black shales. The composition and pressure of these FIs are quantified through in situ Raman spectroscopic analyses. The trapping pressure of the CH4-saturated fluid can reach > 218.03–256.82 MPa, which provides direct evidence for the paleo-overpressure of source rocks during methane generation.
References
Bakker RJ. Adaption of the Bowers and Helgeson (1983) equation of state to the H2O–CO2–CH4–N2–NaCl system. Chem Geol. 1999;154:225–36. https://doi.org/10.1016/S0009-2541(98)00133-8.
Bergman SC, Dubessy J. CO2–CO fluid inclusions in a composite peridotite xenolith: implications for upper mantle oxygen fugacity. Contrib Mineral Petrol. 1984;85:1–13. https://doi.org/10.1007/BF00380216.
Bodnar RJ. Petroleum migration in the Miocene Monterey Formation, California, USA: constraints from fluid-inclusion studies. Mineral Mag. 1990;54:295–304. https://doi.org/10.1180/minmag.1990.054.375.15.
Bodnar RJ. Revised equation and table for determining the freezing point depression of H2O–NaCl solutions. Geochim Cosmochim Acta. 1993;57(3):683–4. https://doi.org/10.1016/0016-7037(93)90378-A.
Bowers TS, Helgeson HC. Calculation of the thermodynamic and geochemical consequence of nonideal mixing in the system H2O–CO2–NaCl on phase relations in geological systems: Equation of state for H2O–CO2–NaCl fluids at high pressures and temperatures. Geochim Cosmochim Acta. 1983;47:1247–75. https://doi.org/10.1016/0016-7037(83)90066-2.
Burruss RC, Cercone KR, Harris PM. Fluid inclusion petrography and tectonic-burial history of the A1 Ali No. 2 well: evidence for the timing of diagenesis and oil migration, northern Oman Foredeep. Geology. 1983;11:567–70. https://doi.org/10.1130/0091-7613(1983)11<567:FIPATH>2.0.CO;2.
Cao J, Yao S, Jin Z, et al. Petroleum migration and mixing in the northwestern Junggar Basin (NW China): constraints from oil-bearing fluid inclusion analyses. Mar Pet Geol. 2006;37:827–46. https://doi.org/10.1016/j.orggeochem.2006.02.003.
Chen Y, Wang X, Bodnar RJ. UV Raman spectroscopy of hydrocarbon-bearing inclusions in rock salt from the Dongying sag, eastern China. Org Geochem. 2016;101:63–71. https://doi.org/10.1016/j.orggeochem.2016.08.010.
Chou I-M, Pasteris JD, Seitz JC. High-density volatiles in the system C–O–H–N for the calibration of a laser Raman microprobe. Geochim Cosmochim Acta. 1990;54(3):535–43. https://doi.org/10.1016/0016-7037(90)90350-T.
Chou I-M, Burruss RC, Lu W. Chapter 24—a new optical capillary cell for spectroscopic studies of geologic fluids at pressures up to 100 MPa. Advances in High-Pressure Technology for Geophysical Applications. 2005. https://doi.org/10.1016/B978-044451979-5.50026-0.
Chou I-M, Song Y, Burruss RC. A new method for synthesizing fluid inclusions in fused silica capillaries containing organic and inorganic material. Geochim. Cosmochim. Acta. 2008;72(21):5217–31. https://doi.org/10.1016/j.gca.2008.07.030.
Collins PLF. Gas hydrates in CO2-bearing fluid inclusions and the use of freezing data for estimation of salinity. Econ Geol. 1979;74:1435–44. https://doi.org/10.2113/gsecongeo.74.6.1435.
Cui J, Wang T, Li M, et al. Oil filling history of the Bashituo Oilfield in the Markit Slope, SW Tarim Basin, China. Pet Sci. 2013;10:58–64. https://doi.org/10.1007/s12182-013-0250-0.
Darling RS. An extended equation to calculate NaCl contents from final clathrate melting temperatures in H2O–CO2–NaCl fluid inclusions: implications for P–T isochore location. Geochim Cosmochim Acta. 1991;55:3869–71. https://doi.org/10.1016/0016-7037(91)90079-K.
Dutkiewicz A, Ridley J, Buick R. Oil-bearing CO2–CH4–H2O fluid inclusions: oil survival since the Palaeoproterozoic after high temperature entrapment. Chem Geol. 2003;194:51–79. https://doi.org/10.1016/S0009-2541(02)00271-1.
Fall A, Eichhubl P, Cumella SP, et al. Testing the basin-centered gas accumulation model using fluid inclusion observations: Southern Piceance Basin Colorado. AAPG Bull. 2012;96:2297–318. https://doi.org/10.1306/05171211149.
Fang RH, Li MJ, Lü HT, et al. Oil charging history and pathways of the Ordovician carbonate reservoir in the Tuoputai region, Tarim Basin, NW China. Pet Sci. 2017;14:662–75. https://doi.org/10.1007/s12182-017-0196-8.
Fang J, Chou IM, Chen Y. Quantitative Raman spectroscopic study of the H2–CH4 gaseous system. J Raman Spectrosc. 2018;49(4):710–20. https://doi.org/10.1002/jrs.5337.
Goldstein RH, Reynolds TJ. Systematics of fluid inclusions in diagenetic minerals. SEPM Short Course. 1994;31:199. https://doi.org/10.2110/scn.94.31.
Guo X, Liu K, He S, et al. Petroleum generation and charge history of the northern Dongying Depression, Bohai Bay Basin, China: insight from integrated fluid inclusion analysis and basin modelling. Mar Pet Geol. 2012;32:21–35. https://doi.org/10.1016/j.marpetgeo.2011.12.007.
Hansen SB, Berg RW, Stenby EH. Raman spectroscopic studies of methane–ethane mixtures as a function of pressure. Appl Spectrosc. 2001;55(6):745–9. https://doi.org/10.1366/0003702011952442.
Hollister L, Crawford M. Fluid inclusions, applications to petrology (a short course sponsored by the Mineralogical Association of Canada and held immediately prior to the 1981 annual meeting in Calgary, Alberta, May 7th–10th, 6. Mineralogical Association of Canada; 1981. https://doi.org/10.1016/0016-7037(82)90175-2.
Hunt JM. Petroleum geochemistry and geology. New York: W.H. Freeman and Company; 1996. https://doi.org/10.1016/0009-2541(80)90124-2.
Jager MD, Sloan ED. The effect of pressure on methane hydration in pure water and sodium chloride solutions. Fluid Phase Equilib. 2001;185(1):89–99. https://doi.org/10.1016/S0378-3812(01)00459-9.
Jia C, Zheng M, Zhang Y. Unconventional hydrocarbon resources in China and the prospect of exploration and development. Pet Explor Dev. 2012;39:129–36. https://doi.org/10.1016/S1876-3804(12)60026-3(in Chinese).
Jiao K, Ye Y, Liu S, et al. Characterization and evolution of nanoporosity in superdeeply buried shales: a case study of the Longmaxi and Qiongzhusi Shales from MS Well #1, North Sichuan Basin, China. Energy Fuels. 2018;32:191–203. https://doi.org/10.1021/acs.energyfuels.7b02932.
Jin Z, Hu Z, Gao B, et al. Controlling factors on the enrichment and high productivity of shale gas in the Wufeng-Longmaxi Formations, southeastern Sichuan Basin. Earth Sci Front. 2016;23:1–10. https://doi.org/10.13745/j.esf.2016.01.001(in Chinese).
Khorasani GK. Novel development in fluorescence microscopy of complex organic mixtures: applications in petroleum geochemistry. Org Geochem. 1987;11:157–68. https://doi.org/10.1016/0146-6380(87)90019-2.
Kim SB, Hammaker RM, Fateley WG. Calibrating Raman spectrometers using a neon lamp. Appl Spectrosc. 1986;40(3):412–5. https://doi.org/10.1366/0003702864509231.
Li YJ, Liu H, Zhang LH, et al. Lower limits of evaluation parameters for the lower Paleozoic Longmaxi shale gas in southern Sichuan Province. Sci China Earth Sci. 2013;56:710–7. https://doi.org/10.1007/s11430-013-4579-4.
Li R, Qin X, Dong S, et al. Characteristics of hydrocarbon fluid inclusions and their significance for evolution of petroleum systems in the Dabashan foreland, Central China. Acta Geol Sin (English Edition). 2015;89:861–75. https://doi.org/10.1111/1755-6724.12484.
Li YF, Sun W, Liu XW, et al. Study of the relationship between fractures and highly productive shale gas zones, Longmaxi Formation, Jiaoshiba area in eastern Sichuan. Pet Sci. 2018;15:498–509. https://doi.org/10.1007/s12182-018-0249-7.
Lin F, Sum AK, Bodnar RJ. Correlation of methane Raman ν1 band position with fluid density and interactions at the molecular level. J Raman Spectrosc. 2007;38(11):1510–5. https://doi.org/10.1002/jrs.1804.
Liu DH, Dai JX, Xiao XM, et al. High density methane inclusions in Puguang gasfield: discovery and T–P genetic study. Chin Sci Bull. 2009;54:4714–23. https://doi.org/10.1007/s11434-009-05.
Lu WJ, Chou I-M, Burruss RC, et al. A unified equation for calculating methane vapor pressures in the CH4–H2O system with measured Raman shifts. Geochim Cosmochim Acta. 2007;71(16):3969–78. https://doi.org/10.1016/j.gca.2007.06.004.
Lüders V, Plessen B, di Primio R. Stable carbon isotopic ratios of CH4–CO2-bearing fluid inclusions in fracture-fill mineralization from the Lower Saxony Basin (Germany)—a tool for tracing gas sources and maturity. Mar Pet Geol. 2012;30:174–83. https://doi.org/10.1016/j.marpetgeo.2011.10.006.
Mernagh TP, Wilde AR. The use of the laser Raman microprobe for the determination of salinity in fluid inclusions. Geochim Cosmochim Acta. 1989;53(4):765–71. https://doi.org/10.1016/0016-7037(89)90022-7.
Munz IA. Petroleum inclusions in sedimentary basins: systematics, analytical methods and applications. Lithos. 2001;55:195–212. https://doi.org/10.1016/S0024-4937(00)00045-1.
National (1980) 1:200,000 Geological Map (H-48-23) (Paper Edition). Sichuan Geology and Mineral Bureau Regional Geological Survey Team, China Geological Survey. 1980.
NIST Chemistry WebBook. https://webbook.nist.gov/chemistry. Accessed 18 Sept 2019.
Pasteris JD, Wopenka B, Seitz JC. Practical aspects of quantitative laser Raman microprobe spectroscopy for the study of fluid inclusions. Geochim Cosmochim Acta. 1988;52:979–88. https://doi.org/10.1016/0016-7037(88)90253-0.
Petrychenko OY, Peryt TM, Chechel EI. Early Cambrian seawater chemistry from fluid inclusions in halite from Siberian evaporates. Chem Geol. 2005;219:149–61. https://doi.org/10.1016/j.chemgeo.2005.02.003.
Pironon J, Thiéry R, Ougougdal MA, et al. FT-IR measurements of petroleum fluid inclusions: methane, n-alkanes and carbon dioxide quantitative analysis. Geofluids. 2001;1:2–10. https://doi.org/10.1046/j.1468-8123.2001.11002.x.
Ran B, Liu S, Jansa L, et al. Reservoir characteristics and preservation conditions of Longmaxi shale in the Upper Yangtze Block, South China. Acta Geol Sin (English Edition). 2016;90:2182–205. https://doi.org/10.1111/1755-6724.13030.
Roedder E. Fluid inclusions; reviews in mineralogy. Mineral Soc Am. 1984;12:1–644. https://doi.org/10.1016/0016-7037(85)90299-6.
Roedder E, Bodnar RJ. Geologic pressure determinations from fluid inclusion studies. Ann Rev Earth Planet Sci. 1980;8:263–301. https://doi.org/10.1146/annurev.ea.08.050180.001403.
Schmidt C. Raman spectroscopic study of a H2O + Na2SO4 solution at 21–600°C and 0.1 MPa to 1.1 GPa, Relative differential Raman scattering cross sections and evidence of the liquid–liquid transition. Geochim Cosmochim Acta. 2009;73:425–37. https://doi.org/10.1016/j.gca.2008.10.019.
Seitz JC, Pasteris JD, Chou I-M. Raman spectroscopic characterization of gas mixtures; I, Quantitative composition and pressure determination of CH4, N2 and their mixtures. Am J Sci. 1993;293:297–321. https://doi.org/10.2475/ajs.293.4.297.
Seitz JC, Pasteris JD, Chou I-M. Raman spectroscopic characterization of gas mixtures. II. Quantitative composition and pressure determination of the CO2–CH4 system. Am J Sci. 1996;296(6):577–600. https://doi.org/10.2475/ajs.296.6.577.
Shang L, Chou I-M, Burruss RC, et al. Raman spectroscopic characterization of CH4 density over a wide range of temperature and pressure. J Raman Spectrosc. 2015;45(8):696–702. https://doi.org/10.1002/jrs.4529.
Spencer CW. Hydrocarbon generation as a mechanism for overpressing in Rocky Mountain Region. AAPG Bull. 1987;71(4):368–88. https://doi.org/10.1306/94886eb6-1704-11d7-8645000102c1865d.
Thieu V, Subramanian S, Colgate SO, et al. High-pressure optical cell for hydrate measurements using Raman spectroscopy. Ann N Y Acad Sci. 2000;912(1):983–92. https://doi.org/10.1111/j.1749-6632.2000.tb06853.x.
Tissot BP, Welte DH. Petroleum formation and occurrence. New York: Springer; 1984. https://doi.org/10.1007/978-3-642-87813-8.
Ungerer P, Behar E, Discamps D. Tentative calculation of the overall volume expansion of organic matter during hydrocarbon genesis from geochemistry data: Implications for primary migration. In: Bjorøy M, et al., editors. Advances in organic geochemistry. Chichester: Wiley; 1983. p. 129–35.
Thiéry R, Pironon J, Walgenwitz F, et al. PIT (Petroleum Inclusion Thermodynamic): a new modeling tool for the characterization of hydrocarbon fluid inclusions from volumetric and microthermometric measurements. J Geochem Explor. 2000;69-70:701–4. https://doi.org/10.1016/s0375-6742(00)00085-6get.
Walter BF, Steele-MacInnis M, Markl G. Sulfate brines in fluid inclusions of hydrothermal veins: compositional determinations in the system H2O–Na–Ca–Cl–SO4. Geochim Cosmochim Acta. 2017;209:184–203. https://doi.org/10.1016/j.gca.2017.04.027.
Wan Y, Wang X, Hu W, et al. Raman Spectroscopic observations of the ion association between Mg2+ and SO42− in MgSO4-saturated droplets at temperatures of ≤ 380°C. J Phys Chem A. 2015;119(34):9027–36. https://doi.org/10.1021/acs.jpca.5b02938.
Wan Y, Wang X, Hu W, et al. In situ optical and Raman spectroscopic observations of the effects of pressure and fluid composition on liquid–liquid phase separation in aqueous cadmium sulfate solutions (≤ 400°C, 50 MPa) with geological and geochemical implications. Geochim Cosmochim Acta. 2017;211:133–52. https://doi.org/10.1016/j.gca.2017.05.020.
Wang X, Chou I-M, Hu W, et al. Raman spectroscopic measurements of CO2 density: experimental calibration with high-pressure optical cell (HPOC) and fused silica capillary capsule (FSCC) with application to fluid inclusion observations. Geochim Cosmochim Acta. 2011;75(14):4080–93. https://doi.org/10.1016/j.gca.2011.04.028.
Wang R, Hu Z, Long S, et al. Differential characteristics of the Upper Ordovician-Lower Silurian Wufeng-Longmaxi shale reservoir and its implications for exploration and development of shale gas in/around the Sichuan Basin. Acta Geol Sin (English Edition). 2019;93:520–35. https://doi.org/10.1111/1755-6724.13875.
Wopenka B, Pasteris JD. Limitations to quantitative analysis of fluid inclusions in geological samples by laser Raman microprobe spectroscopy. Appl Spectrosc. 1986;40(2):144–51. https://doi.org/10.1366/0003702864509592.
Wopenka B, Pasteris JD. Raman intensities and detection limits of geochemically relevant gas mixtures for the laser Raman microprobe. Anal Chem. 1987;59:2165–70. https://doi.org/10.1021/ac00144a034.
Xi B, Shi W, Jiang H, et al. Synthesis of N2–CH4 and N2–CO2 gas mixtures as an online standard and determination of their Raman quantification factors of CH4 and CO2. Rock Miner Anal. 2014;33(05):655–60 (in Chinese).
Xiong Y, Zhang H, Geng X, et al. Thermal cracking of n-octadecane and its geochemical significance. Chin Sci Bull. 2004;49(Suppl. I):79–83. https://doi.org/10.1007/BF02890457.
Yang L, Ran B, Han YY, et al. Sedimentary environment controls on the accumulation of organic matter in the Upper Ordovician Wufeng-Lower Silurian Longmaxi mudstones in the Southeastern Sichuan Basin of China. Pet Sci. 2019;16:44–57. https://doi.org/10.1007/s12182-018-0283-5.
Zou C, Dong D, Wang Y, et al. Shale gas in China: characteristics, challenges and prospects (I). Petr Explor Dev. 2015;42:689–701. https://doi.org/10.1016/S1876-3804(15)30072-0(in Chinese).
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant Nos. 41922023 and 41830425) and the Fundamental Research Funds for the Central Universities (Grant Nos. 020614380056 and 020614380078). We thank Mr Qianqian Feng from China University of Petroleum (Beijing) for providing the burial history diagram.
Author information
Authors and Affiliations
Corresponding author
Additional information
Handled by Associate Editor Jian Cao
Edited by Jie Hao
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Qiu, Y., Wang, XL., Liu, X. et al. In situ Raman spectroscopic quantification of CH4–CO2 mixture: application to fluid inclusions hosted in quartz veins from the Longmaxi Formation shales in Sichuan Basin, southwestern China. Pet. Sci. 17, 23–35 (2020). https://doi.org/10.1007/s12182-019-00395-z
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12182-019-00395-z