
Abstract
In this study, the influence of CO2 on the rheology of silica poor and K-rich melts from highly explosive eruptions from the Colli Albani Volcanic District (Italy) (CAVD) is measured for the first time. The investigated melts range from foidite to tephri-phonolite to tephrite from the CAVD to a phonolite from the Vesuvius (Italy) with CO2 concentrations up to 0.50 wt%. Viscosity and calorimetric measurements are performed in the glass transition range Tg between 600 and 780 °C. Although nominally anhydrous, the investigated melts contain H2O concentrations up to 0.23 wt%. The data exhibit a decrease in viscosity of approx. 100.40 Pa s for the phonolitic composition with ~ 0.07 wt% CO2 and a Tg reduced by approx. 14 °C. For the tephritic composition, Tg is approx. 5 °C lower and has a viscosity reduced by 100.25 Pa s for the sample containing ~ 0.5 wt% CO2. Calorimetric measurements of the tephri-phonolite show lowered onset of Tg by approx. 6 °C for the melt with ~ 0.11 wt% CO2 and Tg of the foidite appears not to be influenced by a CO2 concentration of ~ 0.37 wt% CO2. However, these tephri-phonolitic and foiditic melts foamed during calorimetric measurements preventing a reliable measurement. It would appear that most of this overall drop in viscosity is caused by the small amounts of H2O in the melts with CO2 slightly reducing the viscosity or having no effect on viscosity. Additionally, it is shown that the reduction in viscosity decreases with an increasing degree of the depolymerisation for the investigated melts. Consequently, the explosive style of the CAVD eruptions is mainly caused by crystals and bubbles which form and rise during magma storage and ascent which increases the magma viscosity whereas the CO2 in the melt slightly reduces the viscosity.
Similar content being viewed by others

Introduction
Rheology of melts plays a key role in magmatic processes. It influences mass transfer, crystallisation processes, nucleation, forming and exsolution of bubbles, the speed of ascent in the volcanic vent and finally the eruptive style. Rheology is affected by melt composition and its volatile content, temperature, pressure, crystal and bubble content as well as oxygen fugacity (i.e. Webb et al. 2007; Chevrel et al. 2013; Di Genova et al. 2014; Campagnola et al. 2016; Klein et al. 2018; Kolzenburg et al. 2018). Among the volatile components in magmatic systems, CO2 is the most important one after H2O (Nowak et al. 2003). In many geological settings with mainly explosive volcanic eruptions, magmas get “flushed” with CO2 by the entrainment of a CO2-rich gas from deeper levels of the magmatic systems or by the assimilation of a CO2-rich host material, i.e. the Colli Albani Volcanic District, Italy (Freda et al. 2011), Mt. Somma-Vesuvius, Italy (Bruno et al. 1998), Popocatépetl volcano, Mexico (Goff et al. 2001) or Merapi volcano, Indonesia (Troll et al. 2012). An enrichment of CO2 in magma results in changes in the rheological behaviour with a decrease in density, changes in chemistry and mineral assemblage and the formation of bubbles and/or crystals. Hence, CO2 is often assumed to trigger volcanic eruptions (Blythe et al. 2015; Caricchi et al. 2018). Blythe et al. (2015) pointed out that the viscosity of a melt strongly determines the carbonate assimilation rate when a magma interacts with the carbonate wall rock. In contrast, the influence of CO2 on melt viscosity is poorly investigated as shown below.
Adding CO2 to polymerised liquids such as NaAlSi3O8 (Brearley and Montana 1989) or KAlSi3O8 (White and Montana 1990) decreases the viscosity at high pressure. In contrast, Brearley and Montana (1989) detected no influence of CO2 on the viscosity of the more depolymerised liquid NaCaAlSi2O7. Viscosity studies on a K-rich silicate melt by Bourgue and Richet (2001) exhibit a reducing effect of CO2 on the viscosity within the first 3 wt% CO2 added. They also measured a linearly decreasing density of the melt with increasing CO2 concentration at room temperature. Morizet et al. (2007) inferred viscosity from calorimetric measurements of the glass transition temperature Tg of CO2 bearing jadeitic and phonolitic melts. They concluded there is little or no influence of dissolved CO2 on viscosity at temperatures near the glass transition because they saw no systematic variations in Tg. Seifert et al. (2013) did experiments on the relaxation times for basaltic, phonolitic and rhyolitic glasses and showed a reduction in relaxation time with increasing CO2 content for the basaltic melt but no changes for the other compositions. As a consequence, they deduce CO2 decreases the viscosity of basaltic melts but does not affect the other SiO2 rich melts. Di Genova et al. (2014) investigated rheological features of natural hydrous and H2O-CO2-bearing trachybasaltic and latitic compositions. They showed a decrease in viscosities for volatile bearing melts and found a stronger decrease in H2O-CO2-bearing melts than for hydrous ones. The glass transition temperature was reduced by ~ 25 K with the addition of any amount of CO32− up to 1900 ppm CO2. In contrast, the heat capacities are not affected by those volatiles. A study by Robert et al. (2015) on a remelted natural basalt confirms the effect of CO2 slightly reducing viscosity in the presence of up to 1 wt% H2O.
The present study investigates the influence of CO2 on rheological properties of three synthetic equivalents of the Colli Albani Volcanic District (CAVD) and a fourth sample equivalent to the “white pumice” phonolite from the Vesuvius AD 79 eruption (WPVe series) (Iacono Marziano et al. 2007). To minimise crystallisation and structural changes during experimental runs, all CAVD glasses are made with half the amount of Fe in the natural sample. Fe2+ is replaced by Mg2+ and Fe3+ by Al3+. Glasses of the CAVD are of the foiditic composition from the Pozzolane Rosse event after Freda et al. (2011) (PR-0.5Fe series) and a tephri-phonolitic one from the Pozzolane Nere event after Campagnola et al. (2016) (PN-0.5Fe series). A further tephritic glass is synthesised by subtracting 4 mol% KAlSi2O6 from the Pozzolane Rosse composition (PR-Leu-0.5Fe series) to investigate the rheology of the residual PR-0.5Fe melt after crystallisation of leucite which is a characteristic phenocryst in the pyroclastic products of the CAVD (i.e. Giordano et al. 2006; Freda et al. 2011).
Methods
Sample preparation
Volatile-free base glasses were made of powdered and previously dried metal oxides and carbonates. They were melted at 1600 °C at ambient pressure and atmosphere for 1 h and subsequently grinded and mixed for 20 min. This procedure was repeated before the glass was finally melted for the third time for 4 h. The glasses were quenched by dipping the crucible with the melt in water at ambient temperature. The base glasses are than powdered and dried at 500 °C for 20 h to avoid adsorption of H2O. The sample powder is filled into Au75Pd25 capsules and welded at approx. 500 °C. For the CO2- bearing melts, the powdered base glass is mixed with silver oxalate Ag2C2O4 as the source of CO2 loaded into the capsules and welded at room temperature. The experiments are performed in an internally heated pressure vessel (IHPV) with an intrinsic oxygen fugacity \(f_{{{\text{O}}_{{\text{2}}} }}\) = NNO + 3 at 1250 °C and approx. 250 MPa for 46 or 70 h (for a detailed description of the IHPV see Schanofski et al. 2019). The melts are quenched with a cooling rate of ~ 150 °C s−1 previously determined in similar studies (Benne and Behrens 2003).
Density
Based on Archimedes’ principle, the sample is weighed in air and in distilled H2O with a wetting agent to reduce the surface tension of H2O using a Mettler Toledo Excellence XP/XS analyse balance. The calculation of the density ρ follows the equation
with A: sample weight in air, B: sample weight in distilled H2O and \(\rho _{{{\text{H}}_{{\text{2}}} {\text{O}}}}\): density of the H2O at ambient temperature after Bigg (1967). The density of every glass is measured 10 times.
Fourier transform infrared spectroscopy (FTIR)
Contents of CO2 and H2O are determined by FTIR spectroscopy in the mid-infrared (MIR) range of 1200–4000 cm−1 and in the near-infrared (NIR) range of 2000–6000 cm−1, respectively. Measurements are done with a Bruker Hyperion 3000 IR microscope and a coupled Bruker Vertex 70 FTIR spectrometer. A tungsten lamp, a MCT detector cooled by liquid nitrogen and a KBr beam splitter together with a Globar light source and a CaF2 beam splitter are used to collect the MIR and NIR spectra, respectively. 100 scans in transmission with 4 cm−1 spectral resolution are collected for each spectrum with a spot size of 50 × 50 µm. Samples are double polished and have a thickness of 100–190 µm for the MIR spectra and 200–250 µm for the NIR spectra.
Colorimetric micro-determination of the iron species
The colorimetric method of Wilson (1960) and Schuessler et al. (2008) was used to determine the Fe speciation. Each sample is powdered and 5–15 mg are dissolved in hydrofluoric acid (HF). Upon addition of 2:2′bipyridyl to the solution, a reddish Fe(II)-bipyridyl-complex is built by the Fe2+. This complex has an intensive absorption band at ~ 523 nm in an UV/VIS spectrum. Hydroxylamine hydrochloride (H4NOCl) is then used to reduce Fe3+ to Fe2+ to measure the total Fe content Fetot. Comparison of the intensities at ~ 523 nm of the unreduced and reduced solutions provides the ratio of Fe2+ to the total Fe content Fe2+/Fetot. The UV/VIS measurements are performed at an AvaSpec-UV/VIS/NIR spectrometer from Avantes and the AvaSoft software Version 8.3.
Micropenetration technique
Viscosity η is measured using the micropenetration technique with a Netzsch TMA 402 dilatometer in the range 108.5–1013.5 Pa s. The calibration of the temperature is done with the melting points of Bi (268.02 °C), Zn (417.02 °C), Al (659.02 °C), NaCl (800.05 °C) and Ag (951.09 °C). The DGG1, a Na2O-CaO-SiO2 float glass from the Deutsche Glastechnische Gesellschaft, is measured as a standard glass to determine the precision of the dilatometer. A total error of ± 0.06 log10 Pa s (1σ) is assessed for the measurements. Samples with parallel surfaces are double polished and have a thickness of 1.5 mm. A sphere of a single crystal of Al2O3 with a diameter of 1 mm is forced into the discs with 0.1–1.5 N. Depending on the expected viscosity, the measurements are performed for 30 min up to 6 h in a temperature range of 600–780 °C. The penetration rate of the sphere into the glass disc is used for the calculation of the viscosity by:
with F: the applied force, t: time, r: radius of the indent sphere, l: distance of indent (Pocklington 1940; Tobolsky and Taylor 1963; Dingwell et al. 1992). As this equation assumes that both the thickness of the sample and the radius of the sphere is much greater than the penetration distance, both F and t are chosen such that l < 80 µm in all of the measurements.
Differential scanning calorimetry (DSC)
Heat capacities are measured with the Netzsch DSC 404C differential scanning calorimeter. The samples are heated to the approximate temperature at which the viscosity is 109 Pa s (Tg9) which is in the temperature range of 730–800 °C, depending on the chemical composition. Measurements are performed with a constant heating- and cooling rate of 20 °C min−1. To erase the thermal history of the glasses, only the second calorimetric run of each sample is used for the determination of the glass transition temperature Tg. Measurements are done in a Pt crucible against an empty Pt crucible and calibrated with a single sapphire crystal with the data of Robie et al. (1978) from 20 to 1000 °C. Measured discs of the WPVe and PR-Leu-0.5Fe series as well as the CO2-free samples of the PN-0.5Fe and PR-0.5Fe series were double polished with parallel faces. During their first calorimetric runs, the discs of CO2-bearing samples of the PN-0.5Fe and PR-0.5Fe series foamed and raised the lid of the crucible. Hence, these samples were subsequently crushed to coarse fragments before their measurements.
The configurational heat capacity Cpconf is determined from the scanning data and is the difference between the relaxed heat capacity of the melt and that of the glass at the same temperature just above the glass transition (Cpconf = Cpl − Cpg).
The liquid heat capacity (Cpl) is directly given by the heat capacity curve. To extrapolate the heat capacity of the glass (Cpg) to a higher temperature, the unrelaxed glass data are fitted by the Maier–Kelley equation:
with a, b, c as parameters and T: temperature in Kelvin (Maier and Kelley 1932). The unrelaxed Cpg is extrapolated to approx. 150 °C to the temperature at which the relaxed Cpl has been determined.
Results
The results of the electron microprobe analysis of the chemical compositions of the investigated melts are listed in Table 1 as well as the chemical compositions calculated with the atomic analysis of the electron microprobe and the Fe ratio Fe2+/Fetot received from the colorimetric micro-determination. All melts are metaluminous by the definition: (Na2O + K2O) < Al2O3 < (CaO + MgO + Na2O + K2O) in mole fraction (Shand 1927). Fe2+/Fetot ranges from 0.33 (PR-0.5Fe_0.2) to 0.51 (samples within the PN-0.5Fe series) (Table 1). The degree of polymerisation of the melt structure is given by γ = (Na2O + K2O + CaO + MgO + FeO)/(Na2O + K2O + CaO + MgO + FeO + Al2O3 + Fe2O3) in mole fraction (Toplis et al. 1997; Webb et al. 2007) and seen in Table 1. The most depolymerised melts are those of the PR-Leu-0.5Fe series (γ = 0.72), followed by the PR-0.5Fe series (γ = 0.68‒0.70) and the PN-0.5Fe series (γ = 0.59‒0.60). The melts of the WPVe series have the highest degree of polymerisation (γ = 0.55). All melts are nominally anhydrous (H2O < 0.50 wt%). The WPVe and PR-Leu-0.5Fe series are crystal free. The PN-0.5Fe and PR-0.5Fe series contain up to 4 vol% crystals with a mean aspect ratio (= major axes/minor axes) of 1.29 and grain sizes from 2 to 4 µm (Table 2) that formed during the IHPV runs. All CO2-free glasses are bubble free. The CO2-bearing samples of the WPVe and the PN-0.5Fe series contain up to 3 vol% spherical bubbles (Table 2). Raman spectroscopy identified the gases in the bubbles as molecular CO2 with characteristic peaks at 1286 and 1390 cm−1 (Schrötter and Klöckner 1979) (for the Raman spectra see Fig. A1 in Appendix A). The crystals as well as the bubbles are unconnected in the crystal and bubble bearing glasses. All glasses are checked for homogeneity by the FTIR. The CO2-bearing melts of the PR-0.5Fe series showed little variations of the CO2 concentration within the samples. Thus, the CO2 concentration of these melts are determined after their viscosity and heat capacity measurements and given in Table 2.
Density
The densities are shown in Table 2. The WPVe series has the lowest densities with 2.47–2.48 g cm−3, followed by the PN-0.5Fe series with 2.57–2.60 g cm−3. The PR-0.5Fe and the PR-Leu-0.5Fe series have similar densities, ranging from 2.70–2.73 to 2.71–2.74 g cm−3, respectively. Within each series of the investigated melts the density appears to decrease with increasing CO2 concentration (Fig. 1A). This decrease in density is, however, within the error of the density of the volatile-free glasses.

A Densities of the investigated melts as a function of CO2 concentration; B Tg12 vs CO2 concentration for the WPVe and PR-Leu-0.5Fe series; C Be/Sconf vs CO2 content for the WPVe and PR-Leu-0.5Fe series. Symbols for the melt series are the same for the three panels
FTIR spectroscopy
The relation between the concentration of a species and the IR-absorbance is given by the Lambert–Beer-law:
with ci: concentration of the species i in wt%, Mi: molar mass of the species i, Aj: peak height of the absorbance of the band j, d: thickness of the sample in cm, ρ: density of the sample in g l−1, ε: absorption coefficient in l mol−1 cm−1 (i.e. Stolper 1982; Thibault and Holloway 1994; Behrens et al. 2009).
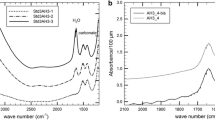
The height of the FTIR-absorption bands related to the carbon species is determined by subtracting the spectrum of the CO2-free glass from that of the CO2-bearing sample scaled to the same thickness. As shown in Fig. 2A, the residual spectrum of the PR-Leu-0.5Fe series shows a doublet at approx. 1510 and 1425 cm−1, caused by the asymmetric stretching vibration of CO32− (Blank and Brooker 1994; Botcharnikov et al. 2006). To quantify the concentration of the CO32− species, the absorbance at approx. 1425 cm−1 is used since the doublet is assumed to be symmetric (Dixon and Pan 1995) and the absorbance at 1510 cm−1 could be overlapped by a vibration mode of H2O molecules (Botcharnikov et al. 2006) and hence falsify the evaluation of the carbon concentration. For this PR-Leu-0.5Fe series, the composition-dependent absorption coefficient ε1425 is calculated using the absorbance normalised by the thickness of the glass chip and density and the total carbon content determined by the carbon sulphur (CS) analysis (Table 2). The value for ε1425 is 324 ± 12 (2σ) l mol−1 cm−1 (see Fig. B1 in Appendix B) being in good agreement with that calculated for Ca-rich silicate glasses from Dixon and Pan (1995) with ε1425 = 335 l mol−1 cm−1.

A MIR spectra of the CO2-bearing melt PR-Leu-0.5Fe_1.3 (continuous line) and the corresponding CO2-free spectra normalised to the thickness of the CO2-bearing one (dashed line). The residual spectrum is the result of the subtraction of the CO2–bearing minus the CO2-free spectra whose absorbance at ~ 1425 cm−1 is used for the determination of the concentration of CO32−; B MIR spectra of the CO2-bearing melt WPVe_1.1 (continuous line) and the corresponding CO2-free spectra normalised to the thickness of the CO2-bearing one (dashed line). The residual spectrum is the result of the subtraction of the CO2-bearing minus the CO2-free spectra. Besides the CO32− doublet at 1425 and 1510 cm−1 a peak at 2350 cm−1 exists indicating the presence of molecular CO2 in this melt
The FTIR spectra of the WPVe and the PN-0.5Fe series show the doublet of CO32− and an extra peak at approx. 2350 cm−1 as shown in Fig. 2B. This extra peak is caused by the asymmetric stretching vibration of CO2 molecules (Blank and Brooker 1994). Since the Raman spectrum (Fig. A1 in Appendix A) attests to the presence of molecular CO2 in the bubbles, it is assumed that this extra peak arises from the substance in the bubbles. The CO2 species in the presented melts is assumed to be dissolved as CO32− only since Fine and Stolper (1986), Thibault and Holloway (1994), Behrens et al. (2009), Shishkina et al. (2014) as well as Schanofski et al. (2019) detected no molecular CO2 in their bubble-free basaltic, Ca-rich leucititic, phono-tephritic, mafic melts as well as phonolitic and foiditic melts, respectively. Therefore, the CS analysis overestimates the total amount of CO2 in these samples and is not reliable for the determination of ε1425 for these melts (an example of the falsified determination of ε1425 is given in Fig. B2 in Appendix B). Due to the underestimations of the absorption coefficients of the bubble bearing melts and the inhomogeneity in CO2 distribution of the CO2-bearing melts of the PR-0.5Fe series, ε1425 values of 245 l mol−1 cm−1 (Schanofski et al. 2019), 296 l mol−1 cm−1 and 334 l mol−1 cm−1 (calculated after Dixon and Pan 1995) are used for the determination of the CO32− content for the WPVe, PN-0.5Fe and the PR-0.5Fe series, respectively. Determined concentrations of CO32− are listed in Table 2.
In the NIR spectra, weak absorbances at approx. 3550 cm−1 are caused by the fundamental OH stretching mode (Stolper 1982; Ohlhorst et al. 2001; Behrens et al. 2009). A two-tangent baseline is used to determine the peak height at 3550 cm−1. There is no evidence of molecular H2O in the glasses, since there is no absorbance peak at approx. 5200 cm−1 which is in agreement with previous studies (i.e. Stolper 1982; Behrens et al. 2009) who observed the OH− groups to be the predominant H2O species in H2O bearing glasses with total H2O content < 0.5 wt%. For the calculation of the total amount of H2O, values for the absorption coefficients ε3550 are taken from other surveys since they strongly depend on melt composition. Stolper (1982) calibrated an absorption coefficient ε3550 for variable compositions ranging from basaltic to albitic to rhyolitic and found it to be the same within error and is 67.0 ± 6.7 l*mol−1*cm−1. Behrens et al. (2009) found ε3550 for a phono-tephritic glass to be 63.9 ± 5.4 l*mol−1*cm−1. For the measurements of the H2O contents of the melts in this study, ε3550 = 67.0 ± 6.7 l*mol−1*cm−1 and ε3550 = 63.9 ± 5.4 l*mol−1*cm−1 are used for the WPVe and PR-0.5Fe series as well as for the PN-0.5Fe and PR-Leu-0.5Fe series, respectively. The melts contain H2O concentrations up to 0.23 wt% (Table 2) due to the reduction of Fe3+ in the samples by hydrogen following the reaction Fe2O3 + H2 \(\to\) 2FeO + H2O (Brearley and Montana 1989; Botcharnikov et al. 2006; Behrens et al. 2009) during sample preparation in the IHPV. An additional possibility for H2O entry into the samples could be adsorbed H2O on the Ag2C2O4 dissolved during the experimental run in the IHPV.
Viscosity
The measured viscosities at a given temperature of all melts are given in the supplementary material (Appendix C). Due to the narrow temperature range applied in this study the statistical analysis needs to fit a straight line through the measured viscosity data which is given by the Arrhenian Equation
with η: viscosity in Pa s, A and B as: fit parameters and T: temperature in Kelvin. All fit parameters are listed in Table 3.
The CO2–bearing samples WPVe_1.1 and WPVe_1.2 contain < 1 vol% spherical bubbles and show bubble growth and rise at temperatures > 700 °C, and therefore these data are not shown.
Einstein (1906) developed a formula for the effect of suspended isolated spheres on melt viscosity:
with ηrel: relative viscosity, ηeff: effective viscosity of the dilute suspension in Pa s, ηmelt: viscosity of the pure suspending liquid in Pa s, f: constant that is 1 for spherical bubbles (Taylor 1932) and φ: bubble fraction.
For the CO2-bearing samples, the influence of the present bubbles on viscosity is < 0.01 log10 Pa s and hence negligible. As seen in Fig. 3A, the viscosities of the CO2-free melts of the WPVe series agree with each other within the measurement error of 0.06 log10 Pa s. At a temperature of 650 °C, the viscosities of the samples WPVe_0.1 and WPVe_0.2 are 12.42 and 12.37 log10 Pa s, respectively. The viscosities of the CO2-bearing samples, having the same CO2 content, are about 0.4 log10 Pa s lower at this temperature, namely 12.01 log10 Pa s for WPVe_1.1 and 11.96 log10 Pa s for WPVe_1.2. The glass transition temperature Tg12 for the CO2-bearing melts is approx. 14 °C lower with respect to that of the CO2-free samples of this series (see Fig. 1B and Table 3).

Viscosity data of all melts investigated in this study as a function of inverse temperature for: A the CO2 free and CO2-bearing melts of the WPVe series, B the CO2 free PN-0.5Fe and PR-0.5Fe melts and C the CO2 free and CO2-bearing melts of the PR-Leu-0.5Fe series. The measured temperature range in °C is given at the top of the figure
The CO2-bearing melts of the PN-0.5Fe and the PR-0.5Fe series disintegrated during heating before reaching the temperature of measurement. It is assumed, that gases in existing or forming bubbles in the PN-0.5Fe series and PR-0.5Fe series, respectively, expand und lead to the fragmentation of these melts. Therefore, only the viscosities of the CO2-free melts are investigated.
As illustrated in Fig. 3B, the viscosity of PN-0.5Fe_0.1 is approx. 0.1 log10 Pa s higher than those of PN-0.5Fe_0.2 over the whole temperature range i.e. at 700 °C the viscosity is 11.25 log10 Pa s and 11.13 log10 Pa s, respectively. The viscosities of the CO2-free samples of the PR-0.5Fe series are additionally shown in Fig. 3B. At a temperature of 700 °C, the viscosity of PR-0.5Fe_0.1 is 10.18 log10 Pa s and that from PR-0.5Fe_0.2 is 10.11 log10 Pa s differing by 0.07 log10 Pa s.
Figure 3C shows the viscosities of the PR-Leu-0.5Fe series. The viscosity data of the CO2-free samples agree with each other, i.e. at 650 °C the viscosities for PR-Leu-0.5Fe_0.1 and PR-Leu-0.5Fe_0.2 are 11.41 and 11.39 log10 Pa s, respectively, what is within the error of measurements. The viscosities of the samples with a CO2 concentration of ~ 0.30 wt% (PR-Leu-0.5Fe_1.1), ~ 0.40 wt% (PR-Leu-0.5Fe_1.2) and ~ 0.50 wt% (PR-Leu-0.5Fe_1.3) are 0.18, 0.22 and 0.25 log10 Pa s lower, respectively, compared to the CO2 free PR-Leu-0.5Fe_0.2. The glass transition temperature Tg12 of the CO2-bearing melts decreases by approx. 5 °C with respect to PR-Leu-0.5Fe_0.2 (see Fig. 1B and Table 3).
Fragility
Using the viscosity data, the fragility m is calculated by
with B: fit parameter from the Arrhenian fit (Eq. 5) and Tg12: temperature at which the viscosity is 1012 Pa s in Kelvin (Toplis et al. 1997; Webb 2011).
As demonstrated in Table 3, the PR-Leu-0.5Fe series has the highest fragilities (38.7–41.2) whereas the fragilities of all other melts for which viscosity measurements are possible, are nearly the same (25.5–28.7).
The Vogel-Fulcher-Tamann Equation (VFT Equation)
with AVFT: constant, BVFT and CVFT: parameters and T: temperature in Kelvin (i.e. Toplis et al. 1997), can be used to force a curve in the fit to the viscosity data. This requires setting the constant AVFT to − 4 (see Kleest et al. (2020) for a discussion). Corresponding to the VFT Equation (Eq. 8), the fragility mVFT is then:
The constant AVFT, the parameters BVFT and CVFT as well as the fragilities using Eq. 9 are given in the supplemental material (Appendix D).
Differential scanning calorimetry (DSC)
The extrapolated parameters of the Maier–Kelley-Fit (Eq. 3) of the studied melts are listed in Table 4 together with the heat capacities of the glasses (cpg) and liquids (cpl) and the configurational heat capacity Cpconf. Cpconf is lowest for the WPVe series (10.35–12.42 J mol−1 K−1), followed by the PN-0.5Fe series (13.76–16.38 J mol−1 K−1). The PR-0.5Fe series has a similar Cpconf (20.70–26.3 J mol−1 K−1) to the PR-Leu-0.5Fe series (25.31–26.93 J mol−1 K−1).
Figure 4 shows is a positive correlation between Cpconf and the degree of depolymerisation expressed by γ which is in agreement with previous studies from Giordano and Russell (2017) and Russell and Giordano (2017) who demonstrated that Cpconf diminishes with increasing SiO2 content. An increase of SiO2 increases the degree of polymerisation and hence a lower re-ordering of the structure in the melt is required at the glass transition range resulting in lower Cpconf.

Configurational heat capacity Cpconf as a function of γ: the higher the degree of polymerisation the lower Cpconf
The fragmentation of the CO2-bearing glasses of the PN-0.5Fe and PR-0.5Fe series prevented viscosity measurements, however, the calorimetric data can be used to estimate the viscosities. Scherer (1984) postulated a relation between viscosity in the glass transition range and the cooling-rate during calorimetric measurements:
with η:– viscosity in Pa s, TgOnset: temperature at the beginning of the glass transition range, defined by the extrapolated glassy heat capacity via the Maier–Kelley fit (Eq. 3) and the rapid increase in heat capacity, KOnset: shift factor of 11.3 and q: cooling rate in °C s−1.
KOnset is calculated for each sample for which viscosity data are available. The average of KOnset (øKOnset) for each melt series is used for further investigations of the CO2-bearing melts and is listed in Table 4 as well as TgOnset.
KOnset varies from 11.56 ± 0.05 for the PR-0.5Fe series to 11.86 ± 0.10 for the WPVe series. KOnset is assumed to be independent of the chemical composition (Stevenson et al. 1995). However, Stevenson et al. (1995) argued that KOnset slightly decreases with increasing agpaitic index ((Na2O + K2O)/Al2O3 in mol. fraction). As seen in Fig. 5 showing øKOnset of each series vs SiO2 concentration, øKOnset slightly increases with increasing SiO2 content. Conventionally implicating a more polymerised melt with increasing SiO2 content, this observation agrees with that from Stevenson et al. (1995) treating the agpaitic index as an indication of the degree of polymerisation of a melt.

Average of the shift factor KOnset of each melt series vs SiO2
The CO2-bearing melts of the WPVe and PN-0.5Fe series contain bubbles of < 1 vol% and 3 vol%, respectively. Based on Eq. 6, the influence of bubbles on viscosity is < 0.01 log10 Pa s for the WPVe melts and 0.01 log10 Pa s for the PN-0.5Fe melts which is within the measurement error of 0.06 log10 Pa s.
The calorimetrically determined viscosities are shown in Fig. 6, presented as the difference in log10 η at TgOnset between the CO2-free and the CO2-bearing samples of each series vs CO2 concentration. For the WPVe, the PN-0.5Fe as well as for the PR-Leu-0.5Fe series, the CO2-bearing melts have lower viscosities at TgOnset than the CO2-free melts.

The viscosity differences of the CO2 bearing and CO2-free melts of each series at TgOnset from DSC measurements vs CO2 concentration. Numbers in rectangles next to the intrapolated lines of each melt series are γ as a value of the degree of polymerisation. The viscosity drop decreases with increasing γ and hence with increasing degree of depolymerisation
The viscosity of the WPVe_1.1 sample decreases by 0.39 log10 Pa s and the WPVe_1.2 sample has a viscosity drop of 0.60 log10 Pa s with respect to the CO2-free WPVe_0.1 melt. In contrast to the results of the viscosity measurements, the viscosities of the CO2-bearing melts of the WPVe series differ by 0.21 log10 Pa s at TgOnset.
The viscosity of the CO2-bearing melts with approx. 0.10 to 0.12 wt% CO2 of the PN-0.5Fe series decreases by approx. 0.27 log10 Pa s in comparison to PN-0.5Fe_0.1 at TgOnset.
The PR-Leu-0.5Fe_1.1 sample with a CO2 concentration of ~ 0.30 wt% has a viscosity diminished by 0.26 log10 Pa s and the viscosities of PR-Leu-0.5Fe_1.2 and PR-Leu-0.5Fe_1.3 with an increased CO2 content up to ~ 0.50 wt% CO2 are reduced up to 0.46 log10 Pa s at TgOnset with respect to the PR-Leu-0.5Fe_0.1 sample.
A difference in viscosity of the PR-0.5Fe series is not detectable since the decrease in viscosity is 0.01 and 0.03 log10 Pa s for PR-0.5Fe_1.1 and PR-0.5Fe_1.2 compared to the CO2 free PR-0.5Fe_0.1 melt which is within the error. However, the CO2-bearing melts of the PR-0.5Fe series foamed during DSC measurements resulting in an incorrect evaluation of TgOnest and consequently to an incorrect calculated viscosity at TgOnest. Since the formation of bubbles due to exsolution of volatiles in the melt already occurs during the first calorimetric run, it shifts the glass transition range in the second run to higher temperatures and hence an increased TgOnset and diminishes the resulting differences in viscosity of the CO2-free and CO2-bearing melts.
The Adam-Gibbs theory combines the data from viscosity and DSC measurements and enables the calculation of the configurational entropy Sconf:
with Ae and Be as constants, Tg12: temperature at which the viscosity is 1012 Pa s in Kelvin (calculated using the Arrhenian Fit Eq. 5), T: temperature in Kelvin and Cpconf: configurational heat capacity (Cpconf = Cpl–Cpg; Cpl is directly measured by DSC and Cpg is extrapolated via Eq. 3) (Adam and Gibbs 1965; Richet 1984; Toplis et al. 1997; Webb 2008) which is valid with the assumption of no variations in Sconf at temperatures below Tg12. The parameter Ae defines the viscosity at infinite temperature and is assumed to be constant for all compositions (i.e. Toplis et al. 1997; Toplis 1998; Giordano et al. 2008). In the present study, Ae is chosen to be − 4.00 log10 Pa s following the Maxwell relation τ = η0/G (with τ:– relaxation time, η0 = time-independent Newtonian viscosity and G = elastic shear modulus) with τ0 = 10–14 s (τ0 = vibration frequency at infinite temperature) (Martinez and Angell 2001; Angell et al. 2003) and G∞ = 10 ± 0.5 GPa (G∞ = shear modulus at infinite frequency) (Dingwell and Webb 1989). For a detailed discussion of Ae see Kleest et al. (2020) and references therein.
Equation 11 fits both, the parameter Be and the configurational entropy Sconf and the results are shown in Table 4. Sconf of the WPVe series, the PN-0.5Fe series and the PR-Leu-0.5Fe series are in a similar range (12.49–19.86 J mol−1 K−1), and the PR-0.5Fe series has the highest Sconf (29.07 and 29.27 J mol−1 K−1). The ratio Be/Sconf indicates the changes of the potential energy barrier to viscous flow (Richet 1984; Toplis 1998; Webb 2005) and the smallest unit moving in a melt structure (Toplis 1998). Figure 1C shows Be/Sconf as a function of the CO2 concentration and it is seen that the CO2-bearing melts have a lower Be/Sconf than the CO2-bearing melts which is more pronounced for the WPVe series than for the PR-Leu-0.5Fe series with a drop of 0.51 × 103 and 0.29 × 103 K, respectively. Hence, the CO2-bearing melts needs to overcome a lower energy barrier to viscous flow.
Discussion
As shown in Figs. 1B, 3A and 6, the viscosity of the WPVe series apparently decreases by the addition of CO2. The H2O content of 0.12 wt% in the CO2-free samples rises to 0.17 and 0.18 wt% for WPVe_1.1 and WPVe_1.2, respectively (see Table 2). It is well studied that H2O reduces the viscosity of a melt as discussed below. Giordano et al. (2008) developed a model to predict the melt viscosity at given temperatures depending on melt composition including the H2O concentration (subsequently denominated as GRD08). GRD08 is applied to the phonolitic WPVe series to estimate the effect of H2O on the viscosity. Corresponding to GRD08, an addition of 0.05 and 0.06 wt% H2O lowers the viscosity by approx. 0.4 log10 Pa s, in the low temperature—high viscosity range as investigated in this study. For the WPVe_1.1 melt, the viscosity decrease determined by the micropenetration technique and the viscosity at TgOnset agree with each other and is approx. 0.4 log10 Pa s (Figs. 3A and 6). The viscosities of the WPVe_1.2 melt determined with the micropenetration method plots nearly on those for the WPVe_1.1 melt (Fig. 3A). Corresponding to GRD08, the viscosity drop for WPVe_1.1 and WPVe_1.2 results from the H2O content wherefore the CO2 influence appears to vanish. On the other hand, the viscosity difference at TgOnset (Fig. 6) is approx. 0.6 log10 Pa s for WPVe_1.2 contrasting to 0.4 log10 Pa s based on GRD08 indicating a combined viscosity reducing the effect of both volatiles H2O and CO2. This influence of H2O seems to be more pronounced at lower temperatures demonstrated by the comparison of the micropenetration viscosity data and the viscosity at TgOnset (Figs. 3A and 6).
All CO2-free samples of the PN-0.5Fe series and PR-0.5Fe series contain microcrystals (Table 2), thus the viscosity of the pure liquid should be less than that measured (i.e. Einstein 1906; Roscoe 1952; Vetere et al. 2010). Klein et al. (2018) established a model for the calculation of the relative viscosity ηrel of crystal-bearing melts considering crystal fraction, crystal volume and shape. By means of the model, the decrease in viscosities of the pure liquid for both series is up to 0.06 log10 Pa s for the maximum crystal fraction of 0.04 in this study, what is within the measurement error.
However, the CO2-free melts of both series show differences in their H2O concentrations (Table 2). As mentioned above, H2O affects the viscosity noticeably. Previous studies pointed out a viscosity decreasing effect of H2O that is larger in the low temperature—high viscosity range than in the high temperature—low viscosity range. This viscosity reducing effect decreases with increasing amounts of H2O and is more pronounced for more polymerised melts than for less polymerised melts. At 700 °C and in the range of H2O concentration as presented in this study for the PN-0.5Fe and PR-0.5Fe series, the drop in viscosity is approx. 0.1 log10 Pa s for a shoshonitic melt (Vetere et al. 2007) or up to approx. 0.4 log10 Pa s for melts of the Campi Flegrei (Misiti et al. 2011). Whittington et al. (2000) observed a viscosity decrease of 0.20 log10 Pa s for an anhydrous tephrite (0.01 wt% H2O) by the addition of 0.12 wt% H2O. Robert et al. (2013) observed a viscosity decrease of approx. 3 log10 Pa s from a “dry” (0.01 wt% H2O) to a 0.56 wt% H2O bearing basaltic andesite at 700 °C.
For the CO2-free melts of the PN-0.5Fe series, the H2O contents are approx. the same within the error (Table 2) resulting in similar viscosities (Fig. 3) differing at the very limit of the measurement error. Figure 6 suggests a CO2-driven viscosity drop for this series at TgOnset. Taking into account strong viscosity reducing effect of H2O, it is not clearly predictable how CO2 affects these melts.
The differences in viscosity of the CO2-bearing melts of the PR-0.5Fe series appear to be negligible at TgOnset (Fig. 6). As mentioned above, the foaming of the CO2-bearing melts of the PR-0.5Fe series falsify the results of the calorimetric experiments and consequently, it is not possible to state the effect of the influence of CO2 on this melt composition.
The viscosities of the CO2-free melts of the PR-Leu-0.5Fe series agree with each other (Fig. 3C). As a consequence, an increased amount of 0.01 wt% H2O (Table 2) has no detectable influence on the viscosities of this melt series. A further addition of approx. 0.02 wt% H2O as found in the CO2-bearing melts of the PR-Leu-0.5Fe series (Table 2) is assumed also to have a negligible (within the ± 0.06 log10 Pa s error in measurements) effect on viscosity (Table 2). The viscosities of the CO2-bearing melts with different CO2 concentrations are within the error of each other (Figs. 3C and 6). The presented data indicate a viscosity decreasing effect by the addition of CO2 as seen in Figs. 1B, 3C and 6. However, this apparent viscosity drop grazes the extreme end of the precision of the measurements.
Based on the presented data, CO2 concentrations up to ~ 0.50 wt% result in a slight to negligible decrease in viscosity. The overall observed viscosity drop of the phonolitic, tephri-phonolitic and tephritic melts is mainly caused by their H2O content with this decrease in viscosity increasing when the degree of polymerisation (γ) increases (Fig. 6).
Geological importance for the PR and PN eruptions
Kleest et al. (2020) showed that the viscosities of the volatile-free melts of the Pozzolane Rosse and Pozzolane Nere event are higher than expected. They also demonstrated a viscosity decrease for the PR melt caused by assimilation of carbonate wall rock due to an enrichment in Ca2+, and an additional reduction in melt viscosity following leucite crystallisation.
Based on the results of the present study, an incorporation of small amounts of CO2 do not affect the melt viscosity to a great extent. In contrast, even a small H2O content in the melt results in a detectable decrease in viscosity and hence, is more effective.
During the storage of the parental PR and PN magmas, crystal formation reaches fractions up to 60 vol% documented by pyroclastic products (Freda et al. 2011). Kleest et al. (2020) calculated a resulting viscosity increase up to two orders in magnitude for a leucite content of 56 vol% after a model from Caricchi et al. (2007). Additionally, carbonate assimilation reaches values of 15 wt% (Iacono-Marziano et al. 2007) which clearly exceeds the solubility of CO2 in the PR melt (Schanofski et al. 2019). Consequently, the melt becomes oversaturated in CO2 resulting in the formation of bubbles as recorded in scoria layers with a vesicularity up to 50% (Giordano et al. 2006). Trolese et al. (2017) found emplacement temperatures of ignimbrites from the PR and PN eruptions of approx. 710 °C and 600–630 °C, respectively, and inferred that magma fragmentation starts at temperatures being approx. 100 °C higher. Since these fragmentation temperatures are near the glass transition for the CO2 free PR-0.5Fe and PN-0.5Fe melts, the magma on the top of the vent is highly viscous or nearly rigid due to the large crystal content. It is suggested, that a sudden release of bubbles as a consequence of carbonate assimilation or depressurisation in the subjacent less viscous magma forces the magma plug out of the vent leading to magma fragmentation during the eruption and results in a highly explosive eruptive style. Similar findings are discussed by Freda et al. (2011) and Marra et al. (2009) for the PR and PN event and by Cross et al. (2014) for the Albano Maar volcanism, postulating free CO2 triggering explosive eruptions at the CAVD.
Conclusion
The presented study concerns the viscosities of CO2-bearing phonolitic, tephri-phonolitic, tephritic and foiditic melt compositions. The carbon is incorporated as CO32− in the melts. With the exception for the foiditic melt, for which no data are reliable, the viscosity apparently decreases with the addition of CO2. However, this drop in viscosity is overshadowed by the decrease in viscosity caused by small H2O concentrations in the melts. This effect is more pronounced in the more polymerised melts. Although it is difficult to separate the effect of H2O and CO2 on the viscosity of theses melts, it would appear that CO2 results in a small to negligible decrease in viscosity at these high viscosity conditions.
References
Adam G, Gibbs JH (1965) On the temperature dependence of coorperative relaxation properties in glass-forming liquids. J Chem Phys 43:139–146
Angell CA, Yue Y, Wang L-M, Colpey JRD, Borick S, Mosa S (2003) Potential energy, relaxation, vibrational dynamics and boson peak, of hyperquenched glasses. J Phys: Condensed Matter 15:1051–1068
Behrens H, Misiti V, Freda C, Vetere F, Botcharnikov RE, Scarlato P (2009) Solubility of H2O and CO2 in ultrapotassic melts at 1200 and 1250 °C and pressure from 50 to 500 MPa. Am Mineral 94:105–120
Benne D, Behrens H (2003) Water solubility in haplobasaltic melts. Eur J Mineral 15:803–814
Bigg PH (1967) Density of water in SI units over the range 0–40 °C. Brit J Appl Phys 18:521
Blank JG, Brooker RA (1994) Experimental studies of carbon dioxide in silicate melts: solubility, speciation and stable carbon isotope behaviour. In: Carroll MR, Holloway JR (eds) Volatiles in Magmas, vol 30. Reviews in mineralogy. Mineralogical Society of America, Washington, pp 157–186
Blythe LS, Deegan FM, Freda C, Jolis EM, Masotta M, Misiti V, Taddeucci J, Troll VR (2015) CO2 bubble generation and migration during magma-carbonate interaction. Contrib Mineral Petrol 169:article 42
Botcharnikov RE, Behrens H, Holtz F (2006) Solubility and speciation of C-O-H fluids on andesitic melt at T = 1100–1300°C and P = 200 and 500 MPa. Chem Geol 229:125–143
Bourgue E, Richet P (2001) The effects of dissolved CO2 on the density and viscosity silicate melts: a preliminary study. Earth Planet Sci Lett 193:57–68
Brearley M, Montana A (1989) The effect of CO2 on the viscosity of silicate liquids at high pressure. Geochim Et Cosmochim Acta 53:2609–2616
Bruno PPG, Cippitelli G, Rapolla A (1998) Seismic study of the Mesozoic carbonate basement around Mt. Somma-Vesuvius, Italy. J Volcanol Geotherm Res 84:311–322
Campagnola S, Vona A, Giordano G (2016) Crystallization kinetics and rheology of leucite-bearing tephriphonolite magmas from the Colli Albani volcano (Italy). Chem Geol 424:12–29
Caricchi L, Burlini L, Ulmer P, Gerya T, Vassalli M, Papale P (2007) Non-Newtonian rheology of crystal-bearing magmas and implications for magma ascent dynamics. Earth Planet Sci Lett 264:402–419
Carichhi L, Sheldrake TE, Blundy J (2018) Modulation of magmatic processes by CO2 flushing. Earth Planet Sci Lett 491:160–171
Chevrel MO, Platz T, Hauber E, Baratoux D, Lavallée Y, Dingwell DB (2013) Lava flow rheology: a comparison of morphological and petrological methods. Earth Planet Sci Lett 384:109–120
Cross JK, Tomlinson EL, Giordano G, Smith VC, De Benedetti AA, Roberge J, Manning CJ, Wulf S, Menzies MA (2014) High level triggers for explosive mafic volcanism: Albano Maar, Italy. Lithos 190–191:137–153
Di Genova D, Romano C, Alletti M, Misiti V, Scarlato P (2014) The effect of CO2 and H2O on Etna and Fondo Riccio (Phlegrean Fields) liquid viscosity, glass transition temperature and heat capacity. Chem Geol 377:72–86
Dingwell DB, Webb SL (1989) Structural relaxation in silicate melts and non-Newtonian melt rheology in geological processes. Phys Chem Minerals 16:508–516
Dingwell DB, Knoche R, Webb SL (1992) The effect of B2O3 on the viscosity of haplogranitic liquids. Am Mineral 77:457–461
Dixon JE, Pan V (1995) Determination of the molar absorptivity of dissolved carbonate in basanitic glass. Am Mineral 80:1339–1342
Einstein A (1906) Eine neue Bestimmung der Moleküldimensionen. Annals Phys 19:289–306
Fine G, Stolper E (1986) Dissolved carbon dioxide in basaltic glasses: concentration and speciation. Earth Planet Sci Lett 76:263–278
Freda C, Gaeta M, Giaccio B, Marra F, Palladino DM, Scarlat P, Sottili G (2011) CO2-driven large mafic explosive eruptions: the Pozzolane Rosse case study from the Colli Albani Volcanic District (Italy). Bull Volcanol 73:241–256
Giordano D, Russell JK (2017) The heat capacity if hydrous multicomponent natural melts and glasses. Chem Geol 461:96–103
Giordano G, De Benedetti AA, Diana A, Diano G, Gaudioso F, Marasco F, Miceli M, Mollo S, Cas RAF, Funiciello R (2006) The Colli Albani mafic caldera (Roma, Italy): stratigraphy, structure and petrology. J Volcanol Geotherm Res 155:49–80
Giordano D, Russell JK, Dingwell DB (2008) Viscosity of magmatic liquids: a model. Earth Planet Sci Lett 271:123–134
Goff F, Love SP, Warren RG, Counce D, Obenholzer J, Siebe C, Schmidt SC (2001) Passive infrared remote sensing evidence for large, intermittent CO2 emissions at Popocatépetl volcano, Mexico. Chem Geol 177:133–156
Iacono Marziano G, Schmidt BC, Dolfi D (2007) Equilibrium and disequilibrium degassing of a phonolitic melt (Vesuvius AD 79 “white pumice”) simulated by decompression experiments. J Volcanol Geotherm Res 161:151–164
Kleest C, Webb SL, Fanara S (2020) Rheology of melts from the Colli Albani Volcanic District (Italy): a case study. Contrib Mineral Petrol 175:82
Klein J, Mueller SP, Helo C, Schweitzer S, Gurioli L, Castr JM (2018) An expanded model and application of the combined effect of crystal-size distribution and crystal shape on the relative viscosity of magmas. J Volcanol Geotherm Res 357:128–133
Kolzenburg S, Di Genova D, Giordano D, Hess KU, Dingwell DB (2018) The effect of oxygen fugacity on the theological evolution of crystallizing basaltic melts. Earth Planet Sci Lett 487:21–32
Maier CG, Kelley KK (1932) An equation for the representation of high-temperature heat content data. J Am Chem Soc 54:3243–3246
Marra F, Karner DB, Freda C, Gaeta M, Renne P (2009) Large mafic eruptions on Alban Hills Volcanic District (Central Italy): Chronostratigraphy, petrography and eruptive behaviour. J Volcanol Geotherm Res 179:217–232
Martinez L-M, Angell CA (2001) A thermodynamic connection to the fragility of glass-forming liquids. Nature 410:663–667
Misiti V, Vetere F, Freda C, Scarlato P, Behrens H, Mangiacarpa A, Dingwell DB (2011) A general viscosity model of Campi Flegrei (Italy) melts. Chem Geol 290:50–59
Morizet Y, Nichols ARL, Kohn SC, Brooker RA, Dingwell DB (2007) The influence of H2O and CO2 on the glass transition temperature: insights into the effect of volatiles on magma viscosity. Eur J Mineral 19:657–669
Nowak M, Porbatzki D, Spickenbom K, Diedrich O (2003) Carbon dioxide speciation in silicate melts: a restart. Earth Planet Sci Lett 207:131–139
Ohlhorst S, Behrens H, Holtz F (2001) Compositional dependence of molar absorptivities of near-infrared OH- and H2O bands in rhyolitic to basaltic glasses. Chem Geol 174:5–20
Pocklington HC (1940) Rough measurement of high viscosities. Proc Cambridge Phil Soc 36:507–508
Richet P (1984) Viscosity and configurational entropy of silicate melts. Geochim Et Cosmochim Acta 48:471–483
Robert G, Whittigton AG, Stechern A, Behrens H (2013) The effect of water on the viscosity of a synthetic calc-alkaline basaltic andesite. Chem Geol 346:135–148
Robert G, Knipping JL, Scherbarth S, Robertson TE, Stechern A, Behrens H, Whittington AG (2015) Heat capacity and viscosity of basaltic melts with H2O ± F ± CO2. Chem Geol 418:51–65
Robie RA, Hemingway BS, Fischer JR (1978) Thermodynamic properties of minerals and related substances at 298.15 K and 1 bar (105 Pa) pressure and higher temperatures. US Geol Surv Bull 1452:428–446
Roscoe R (1952) The viscosity of suspensions of rigid spheres. Br J Appl Phys 3:267–269
Russell JK, Giordano D (2017) Modelling configurational entropy of silicate melts. Chem Geol 461:140–151
Scherer GW (1984) Use of the adam-gibbs equation in the analysis of structural relaxation. Corning Glass Works, R&D Division, New York, 14831
Schanofski M, Fanara S, Schmidt BC (2019) CO2-H2O solubility in K-rich phonolitic and leucititic melts. Contrib Mineral Petrol 174:52
Schrötter HW, Klöckner HW (1979) Raman scattering cross sections in gases and liquids. In: Weber A (eds) Raman spectroscopy of gases and liquids. Topics in current physics, vol 11. Springer, Berlin, Heidelberg
Schuessler JA, Botcharnikov RE, Behrens H, Misiti V, Freda C (2008) Oxidation state of iron in hydrous phono-tephritic melts. Am Mineral 93:1493–1504
Seifert R, Malfait WJ, Lerch P, Sanchez-Valle C (2013) Partial molar volume and compressibility of dissolved CO2 in glassese with magmatic compositions. Chem Geol 358:119–130
Shand SJ (1927) The eruptive rocks. Wiley, New York
Shishkina TA, Botcharnikov RE, Holtz F, Almeev RR, Jazwa AM, Jakubiak AA (2014) Compositional and pressure effects on the solubility of H2O and CO2 in mafic melts. Chem Geol 388:112–129
Stevenson RJ, Dingwell DB, Webb SL, Bagdassarov NS (1995) The equivalence of enthalpy and shear stress relaxation in rhyolitic obsidians and quantification of the liquid-glass transition in volcanic processes. J Volcanol Geotherm Res 68:297–306
Stolper E (1982) Water in silicate glasses: an infrared spectroscopic study. Contrib Mineral Petrol 81:1–17
Taylor GI (1932) The viscosity of a fluid containing small drops of another fluid. Proc R Soc London A 138:41–48
Thibault Y, Holloway JR (1994) Solubility if CO2 in a Ca-rich leucitie: effects of pressure, temperature and oxygen fugacity. Contrib Mineral Petrol 116:216–224
Tobolsky AV, Taylor RB (1963) Viscoelastic properties of a simple organic glass. J Phys Chem 67:2439–2442
Toplis MJ (1998) Energy barriers to viscous flow and the prediction of the glass transition temperatures of molten silicates. Am Mineral 83:480–490
Toplis MJ, Dingwell DB, Hess K-U, Lenci T (1997) Viscosity, fragility and configurational entropy of melts along the join SiO2-NaAlSiO4. Am Mineral 82:979–990
Trolese M, Giordano G, Cifelli F, Winkler A, Mattei M (2017) Forced transport of thermal energy in magmatic and phreatomagmatic large volume ignimbrites: paleomagnetic evidence from the Colli Albani volcano, Italy. Earth Planet Sci Lett 478:179–191
Troll VR, Hilton DR, Jolis EM, Chadwick JP, Blythe LS, Deegan FM, Schwarzkopf LM, Zimmer M (2012) Crustal CO2 liberation during the 2006 eruption and earthquake events at Merapi volcano, Indonesia. Geophys Res Lett 39:11
Vetere F, Behrens H, Misiti V, Ventura G, Holtz F, De Rosa R, Deubener J (2007) The viscosity of shoshonitic melts (Vulcanello Peninsula, Aeolian Islands, Italy): Insight on the magma ascent in dikes. Chem Geol 245:89–102
Vetere F, Behrens H, Holtz F, Vilardo G, Ventura G (2010) Viscosity of crystal-bearing melts and its implication for magma ascent. J Mineral Petrol Sci 105:151–163
Webb SL (2005) Structure and rheology of iron-bearing Na2O-Al2O3-SiO2 melts. Eur J Mineral 17:223–232
Webb SL (2008) Configurational heat capacity of Na2O-CaO-Al2O3-SiO2 melts. Chem Geol 256:92–101
Webb SL (2011) Configurational heat capacity and viscosity of (Mg, Ca, Sr, Ba)-Al2O3-SiO2 melts. Eur J Mineral 23:487–497
Webb SL, Banaszak M, Köhler U, Rausch S, Raschke G (2007) The viscosity of Na2O-CaO-Al2O3-SiO2 melts. Eur J Mineral 19:681–692
White B, Montana A (1990) The effect of H2O and CO2 on the viscosisty of sanidin liquid at high pressures. J Geophys Res 95:683–693
Whittington A, Richet P, Holtz F (2000) Water and the viscosity of depolymerized aluminosilicate melts. Geochim Et Cosmochim Acta 64:3725–3736
Wilson AD (1960) The micro-determination of ferrous iron in silicate minerals by a volumetric and a colorimetric method. Analyst 85:823–827
Acknowledgements
The funding of this research is ensured by the Deutsche Forschungsgemeinschaft grant No. WE 1801/15-1. Special thanks go to Dr. Sara Fanara, Dr. Max Schanofski and Dr. Burkhard Schmidt for their support at the IHPV and the FTIR. For technical assistance, we wish to thank Bettina Schlieper-Ludewig for her help with the viscosity, calorimetry and colorimetric measurements and Marina Horstmann for her help at the carbon sulphur analyser. Furthermore, we wish to thank Dr. Andreas Kronz and Jochen Gätjen for their aid at the electron microprobe as well as Andre Petitjean for the tricky sample preparation. And a great thanks to an anonymous reviewer, to Kelly Russell and to Dante Canil for their very constructive recommendations.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Dante Canil.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kleest, C., Webb, S.L. Influence of CO2 on the rheology of melts from the Colli Albani Volcanic District (Italy): foidite to phonolite. Contrib Mineral Petrol 176, 92 (2021). https://doi.org/10.1007/s00410-021-01838-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00410-021-01838-w