APOE4 Genotype Exerts Greater Benefit in Lowering Plasma Cholesterol and Apolipoprotein B than Wild Type (E3/E3), after Replacement of Dietary Saturated Fats with Low Glycaemic Index Carbohydrates

 , ,
, ,
Abstract
:1. Introduction
2. Methods
2.1. RISCK Study Design
2.2. Study Participants
2.3. Intervention Diets and Study Protocol
2.4. Genotyping
2.5. Analytical Methods
2.6. Statistical Analysis
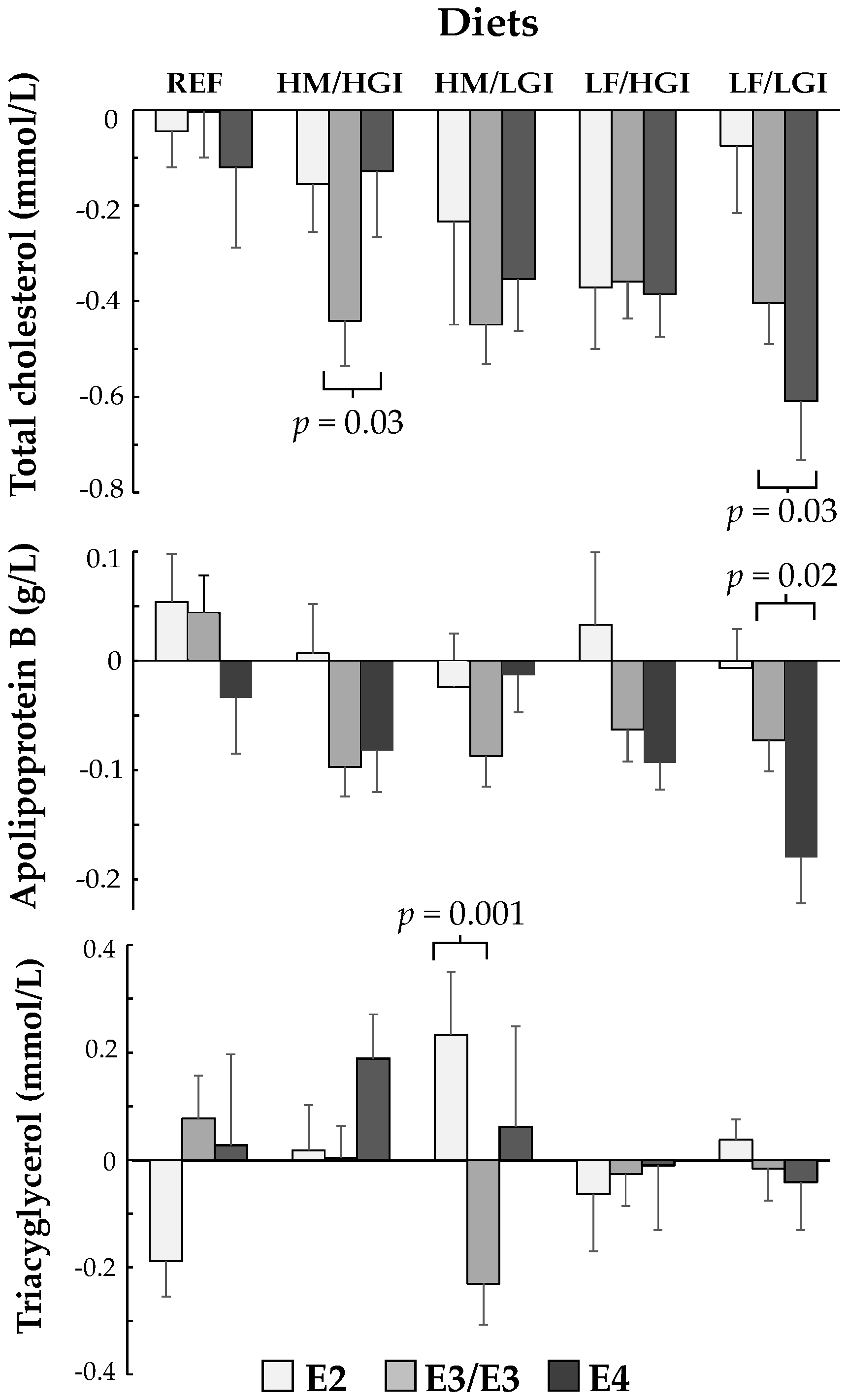
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Walker, C.L. 1983 Nutrition: The changing scene. Implementing the NACNE report. 3. The new British diet. Lancet 1983, 2, 1354–1356. [Google Scholar] [CrossRef]
- Hooper, L.; Martin, N.; Abdelhamid, A.; Davey-Smith, G. Reduction in saturated fat intake for cardiovascular disease. Cochrane Database Syst. Rev. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Cox, C.; Mann, J.; Sutherland, W.; Ball, M. Individual variation in plasma cholesterol response to dietary saturated fat. BMJ 1995, 311, 1260–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Astrup, A.; Dyerberg, J.; Elwood, P.; Hermansen, K.; Hu, F.B.; Jakobsen, M.U.; Kok, F.J.; Krauss, R.M.; Lecerf, J.M.; LeGrand, P.; et al. The role of reducing intakes of saturated fat in the prevention of cardiovascular disease: Where does the evidence stand in 2010? Am. J. Clin. Nutr. 2010, 93, 684–688. [Google Scholar] [CrossRef] [PubMed]
- Siri-Tarino, P.W.; Chiu, S.; Bergeron, N.; Krauss, R.M. Saturated fats versus polyunsaturated Fats versus carbohydrates for cardiovascular disease prevention and treatment. Annu. Rev. Nutr. 2015, 35, 517–543. [Google Scholar] [CrossRef] [PubMed]
- Denke, M.A.; Grundy, S.M. Individual responses to a cholesterol-lowering diet in 50 men with moderate hypercholesterolemia. Arch. Intern. Med. 1994, 154, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Wallace, A.J.; Mann, J.I.; Sutherland, W.H.; Williams, S.; Chisholm, A.; Skeaff, C.M. Variation in plasma cholesterol response to dietary change. Nutr. Metab. Cardiovasc. Dis. 1999, 9, 176–183. [Google Scholar] [PubMed]
- Wilson, P.W.; Schaefer, E.J.; Larson, M.G.; Ordovas, J.M. Apolipoprotein E alleles and risk of coronary disease. A meta-analysis. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 1250–1255. [Google Scholar] [CrossRef] [PubMed]
- Fallaize, R.; Carvalho-Wells, A.L.; Tierney, A.C.; Marin, C.; Kieć-Wilk, B.; Dembińska-Kieć, A.; Drevon, C.A.; DeFoort, C.; Lopez-Miranda, J.; Risérus, U. APOE genotype influences insulin resistance, apolipoprotein CII and CIII according to plasma fatty acid profile in the Metabolic Syndrome. Sci. Rep. 2017, 7, 6274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Povel, C.M.; Boer, J.M.; Imholz, S.; Dollé, M.E.; Feskens, E.J. Genetic variants in lipid metabolism are independently associated with multiple features of the metabolic syndrome. Lipids Health Dis. 2011, 10, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.; Stampfer, M.J.; Liu, S. Meta-analysis: Apolipoprotein E genotypes and risk for coronary heart disease. Ann. Intern. Med. 2004, 141, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.A.; Shah, T.; Prieto, D.; Zhang, W.; Price, J.; Fowkes, G.R.; Cooper, J.; Talmud, P.J.; Humphries, S.E.; Sundstrom, J. Apoprotein E genotype, cardiovascular biomarkers and risk of stroke: Systematic review and meta-analysis of 14015 stroke cases and pooled analysis of primary biomarker data from up to 60 883 individuals. Int. J. Epidemiol. 2013, 42, 475–492. [Google Scholar] [CrossRef] [PubMed]
- Clifton, P.M.; Keogh, J.B. A systematic review of the effect of dietary saturated and polyunsaturated fat on heart disease. Nutr. Metab. Cardiovasc. Dis. 2017, 27, 1060–1080. [Google Scholar] [CrossRef] [PubMed]
- Sarkkinen, E.; Korhonen, M.; Erkkila, A.; Ebeling, T.; Uusitupa, M. Effect of apolipoprotein E polymorphism on serum lipid response to the separate modification of dietary fat and dietary cholesterol. Am. J. Clin. Nutr. 1998, 68, 1215–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campos, H.; D’Agostino, M.; Ordovas, J.M. Gene-diet interactions and plasma lipoproteins: Role of apolipoprotein E and habitual saturated fat intake. Genet. Epidemiol. 2001, 20, 117–128. [Google Scholar] [CrossRef]
- Minihane, A.M.; Khan, S.; Leigh-Firbank, E.C.; Talmud, P.; Wright, J.W.; Murphy, M.C.; Griffin, B.A.; Williams, C.M. Apo E polymorphism and fish oil supplementation in subjects with an atherogenic lipoprotein phenotype. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1990–1997. [Google Scholar] [PubMed]
- Masson, L.F.; McNeill, G.; Avenell, A. Genetic variation and the lipid response to dietary intervention: A systematic review. Am. J. Clin. Nutr. 2003, 77, 1098–1111. [Google Scholar] [CrossRef] [PubMed]
- Paschos, G.K.; Yiannakouris, N.; Rallidis, L.S.; Davies, I.; Griffin, B.A.; Panagiotakos, D.B.; Skopouli, F.N.; Votteas, V.; Zampelas, A. Apolipoprotein E genotype in dyslipidemic patients and response of blood lipids and inflammatory markers to alpha-linolenic acid. Angiology 2005, 56, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Castellanos-Tapia, L.; López-Alvarenga, J.C.; Ebbesson, S.O.; Ebbesson, L.O.; Tejero, M.E. Apolipoprotein E isoforms 3/3 and 3/4 differentially interact with circulating stearic, palmitic, and oleic fatty acids and lipid levels in Alaskan Natives. Nutr. Res. 2015, 35, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Chouinard-Watkins, R.; Conway, V.; Minihane, A.M.; Jackson, K.G.; Lovegrove, J.A.; Plourde, M. Interaction between BMI and APOE genotype is associated with changes in the plasma long-chain-PUFA response to a fish-oil supplement in healthy participants. Am. J. Clin. Nutr. 2015, 102, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Minihane, A.; Jofre-Monseny, L.; Olano-Martin, E.; Rimbach, G. ApoE genotype, cardiovascular risk and responsiveness to dietary fat manipulation. Proc. Nutr. Soc. 2007, 66, 183–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fallaize, R.; Celis-Morales, C.; Macready, A.L.; Marsaux, C.F.; Forster, H.; O’Donovan, C.; Woolhead, C.; San-Cristobal, R.; Kolossa, S.; Hallmann, J.; et al. Food4Me Study. The effect of the apolipoprotein E genotype on response to personalized dietary advice intervention: Findings from the Food4Me randomized controlled trial. Am. J. Clin. Nutr. 2016, 104, 827–836. [Google Scholar] [PubMed]
- Hietaranta-Luoma, H.L.; Åkerman, K.; Tahvonen, R.; Puolijoki, H.; Hopia, A. Using Individual, ApoE genotype-based dietary and physical activity advice to promote healthy lifestyles in Finland—Impacts on cardiovascular risk markers. Open J. Prev. Med. 2015, 5, 206–217. [Google Scholar] [CrossRef]
- Moore, C.; Gitau, R.; Goff, L.; Lewis, F.J.; Griffin, M.D.; Chatfield, M.D.; Jebb, S.A.; Frost, G.S.; Sanders, T.A.; Griffin, B.A.; et al. RISCK Study Group. Successful manipulation of the quality and quantity of fat and carbohydrate consumed by free-living individuals using a food exchange model. J. Nutr. 2009, 139, 1534–1540. [Google Scholar] [PubMed]
- Jebb, S.A.; Lovegrove, J.A.; Griffin, B.A.; Frost, G.S.; Moore, C.S.; Chatfield, M.D.; Bluck, L.J.; Williams, C.M.; Sanders, T.A. RISCK Study Group. Effect of changing the amount and type of fat and carbohydrate on insulin sensitivity and cardiovascular risk: The RISCK (Reading, Imperial, Surrey, Cambridge, and Kings) trial. Am. J. Clin. Nutr. 2010, 92, 748–758. [Google Scholar] [PubMed]
- Vafeiadou, K.; Weech, M.; Altowaijri, H.; Todd, S.; Yaqoob, P.; Jackson, K.G.; Lovegrove, J.A. Replacement of saturated with unsaturated fats had no impact on vascular function but beneficial effects on lipid biomarkers, E-selectin, and blood pressure: Results from the randomized, controlled Dietary Intervention and VAScular function (DIVAS) study. Am. J. Clin. Nutr. 2015, 102, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Davies, I.G.; Graham, J.M.; Griffin, B.A. Rapid separation of LDL subclasses by iodixanol gradient ultracentrifugation. Clin. Chem. 2003, 49, 1865–1872. [Google Scholar] [CrossRef] [PubMed]
- Schiele, F.; De Bacquer, D.; Vincent-Viry, M.; Beisiegel, U.; Ehnholm, C.; Evans, A.; Kafatos, A.; Martins, M.C.; Sans, S.; Sass, C. Apolipoprotein E serum concentration and polymorphism in six European countries: The ApoEurope Project. Atherosclerosis 2000, 152, 475–488. [Google Scholar] [CrossRef]
- Menotti, A.; Lanti, M.; Puddu, P.; Kromhout, D. Coronary heart disease incidence in northern and southern European populations: A reanalysis of the seven countries study for a European coronary risk chart. Heart 2000, 84, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Elosua, R.; Demissie, S.; Cupples, L.A.; Meigs, J.B.; Wilson, P.W.F.; Schaefer, E.J.; Corella, D.; Ordovas, J.M. Obesity modulates the association among APOE genotype, insulin, and glucose in men. Obes. Res. 2003, 11, 1502–1508. [Google Scholar] [CrossRef] [PubMed]
- Torres-Perez, E.; Ledesma, M.; Garcia-Sobreviela, M.P.; Leon-Latre, M.; Arbones-Mainar, J.M. Apolipoprotein E4 association with metabolic syndrome depends on body fatness. Atherosclerosis 2016, 245, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Dallongeville, J.; Lussier-Cacan, S.; Davignon, J. Modulation of plasma triglyceride levels by apoE phenotype: A meta-analysis. J. Lipid Res. 1992, 33, 447–454. [Google Scholar] [PubMed]
- Carvalho-Wells, A.L.; Jackson, K.G.; Gill, R.; Olano-Martin, E.; Lovegrove, J.A.; Williams, C.M.; Minihane, A.M. Interactions between age and ApoE genotype on fasting and postprandial triglyceride levels. Atherosclerosis 2010, 212, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Couture, P.; Archer, W.R.; Lamarche, B.; Landry, N.; Dériaz, O.; Corneau, L.; Bergeron, J.; Bergeron, N. Influences of apolipoprotein E polymorphism on the response of plasma lipids to the ad libitum consumption of a high-carbohydrate diet compared with a high-monounsaturated fatty acid diet. Metabolism 2003, 52, 1454–1459. [Google Scholar] [CrossRef]
- Havel, R.J. Receptor and non-receptor mediated uptake of chylomicron remnants by the liver. Atherosclerosis 1998, 141 (Suppl. 1), S1–S7. [Google Scholar] [CrossRef]
- Bohnet, K.; Pillot, T.; Visvikis, S.; Sabolovic, N.; Siest, G. Apolipoprotein (apo) E genotype and apoE concentration determine binding of normal very low density lipoproteins to HepG2 cell surface receptors. J. Lipid Res. 1996, 37, 1316–1324. [Google Scholar] [PubMed]
- Geisel, J.; Bunte, T.; Bodis, M.; Oette, K.; Herrmann, W. Apolipoprotein E2/E2 genotype in combination with mutations in the LDL receptor gene causes type III hyper-lipoproteinemia. Clin. Chem. Lab. Med. 2002, 40, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Mahley, R.W.; Weisgraber, K.H.; Huang, Y. Apolipoprotein E: Structure determines function, from atherosclerosis to Alzheimer’s disease to AIDS. J. Lipid Res. 2009, S183–S188. [Google Scholar] [CrossRef] [PubMed]
- Jackson, K.G.; Maitin, V.; Leake, D.S.; Yaqoob, P.; Williams, C.M. Saturated fat-induced changes in Sf 60–400 particle composition reduces uptake of LDL by HepG2 cells. J. Lipid Res. 2006, 47, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Calabuig-Navarro, M.V.; Jackson, K.G.; Kemp, C.F.; Leake, D.S.; Walden, C.M.; Lovegrove, J.A.; Minihane, A.M. A randomized trial and novel SPR technique identifies altered lipoprotein-LDL receptor binding as a mechanism underlying elevated LDL-cholesterol in APOE4s. Sci. Rep. 2017, 7, 44119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregg, R.E.; Zech, L.A.; Schaefer, E.J.; Stark, D.; Wilson, D.; Brewer, H.B., Jr. Abnormal in vivo metabolism of apolipoprotein E4 in humans. J. Clin. Investig. 1986, 78, 815–821. [Google Scholar] [CrossRef] [PubMed]
- Demant, T.; Bedford, D.; Packard, C.J.; Shepherd, J. Influence of apolipoprotein E polymorphism on apolipoprotein B-100 metabolism in normolipemic subjects. J. Clin. Investig. 1991, 88, 1490–1501. [Google Scholar] [CrossRef] [PubMed]
- Levy, L.; Tedstone, A. UK Dietary Policy for the Prevention of Cardiovascular Disease. Healthcare 2017, 5, 9. [Google Scholar] [CrossRef] [PubMed]
- U.S. Department of Health and Human Services and U.S. Department of Agriculture. 2015–2020 Dietary Guidelines for Americans, 8th ed.; 2015. Available online: http://health.gov/dietaryguidelines/2015/guidelines/ (accessed on 12 October 2018).
- Praagman, J.; Beulens, J.W.; Alssema, M.; Zock, P.L.; Wanders, A.J.; Sluijs, I.; van der Schouw, Y.T. The association between dietary saturated fatty acids and ischemic heart disease depends on the type and source of fatty acid in the European Prospective Investigation into Cancer and Nutrition-Netherlands cohort. Am. J. Clin. Nutr. 2016, 103, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Siri-Tarino, P.W.; Sun, Q.; Hu, F.B.; Krauss, R.M. Meta-analysis of prospective cohort studies evaluating the association of saturated fat with cardiovascular disease. Am. J. Clin. Nutr. 2010, 91, 535–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chowdhury, R.; Warnakula, S.; Kunutsor, S.; Crowe, F.; Ward, H.A.; Johnson, L.; Franco, O.H.; Butterworth, A.S.; Forouhi, N.G.; Thompson, S.G.; et al. Association of dietary, circulating, and supplement fatty acids with coronary risk: A systematic review and meta-analysis. Ann. Intern. Med. 2014, 160, 398–406. [Google Scholar] [CrossRef]
- Micha, R.; Mozaffarian, D. Saturated fat and cardiometabolic risk factors, coronary heart disease, stroke, and diabetes: A Fresh look at the evidence. Lipids 2010, 45, 893–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Hruby, A.; Bernstein, A.M.; Ley, S.H.; Wang, D.D.; Chiuve, S.E.; Sampson, L.; Rexrode, K.M.; Rimm, E.B.; Willett, W.C.; et al. Saturated Fat as Compared with Unsaturated Fats and Sources of Carbohydrates in Relation to Risk of Coronary Heart Disease: A Prospective Cohort Study. J. Am. Coll. Cardiol. 2015, 66, 1538–1548. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, M.U.; Dethlefsen, C.; Joensen, A.M.; Stegger, J.; Tjønneland, A.; Schmidt, E.B.; Overvad, K. Intake of carbohydrates compared with intake of saturated fatty acids and risk of myocardial infarction: Importance of the glycemic index. Am. J. Clin. Nutr. 2010, 91, 1764–1768. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| E2 Carriers | E3/E3 | E4 Carriers | Global p | |
|---|---|---|---|---|
| Total cholesterol (mmol/L) | 5.36 (0.13) | 5.77 (0.07) | 5.90 (0.09) | 0.001 |
| LDL cholesterol (mmol/L) | 3.15 (0.1) | 3.69 (0.06) | 3.80 (0.08) | 0.0001 |
| HDL cholesterol (mmol/L) | 1.48 (0.06) | 1.41 (0.02) | 1.41 (0.03) | 0.51 |
| Triacylglycerol (mmol/L) | 1.48 (1.31–1.67) | 1.37 (1.30–1.37) | 1.40 (1.28–1.54) | 0.51 |
| Total:HDL-C ratio | 3.83 (0.14) | 4.22 (0.06) | 4.35 (0.09) | 0.002 |
| Apoprotein B (mg/dL) | 84 (3.0) | 99 (2.0) | 108 (3.0) | 0.0001 |
| Apoprotein A-I (mg/dL) | 125 (4.0) | 122 (2.0) | 125 (3.0) | 0.46 |
| Apo B:Apo A-I ratio | 0.69 (0.03) | 0.82 (0.01) | 0.88 (0.03) | 0.0001 |
| Small dense LDL (%) | 22.4 (2.3) | 24.3 (1.1) | 26.6 (1.8) | 0.31 |
| LDL peak density (g/mL) | 1.03 | 1.02 | 1.03 | 0.41 |
| HDL2 (%) | 35.2 (29.9–41.5) | 33.6 (31.0–36.4) | 31.8 (27.6–36.8) | 0.68 |
| Glucose (mmol/L) | 5.67 (5.50–5.84) | 5.67 (5.56–5.77) | 5.74 (5.58–5.90) | 0.60 |
| Insulin (pmol/L) | 61.1 (51.5–72.5) | 58.4 (54.1–63.1) | 60.6 (55.0–66.8) | 0.94 |
| Si (×10−4 mL·µU−1·min−1) | 2.9 (2.4–3.6) | 2.8 (2.6–3.1) | 2.6 (2.3–2.8) | 0.09 |
| AIRg (mL·µU−1·min−1) | 317 (256–394) | 345 (309–385) | 381 (330–441) | 0.29 |
| Disposition index | 978 (745–1282) | 994 (885–1117) | 1151 (984–1346) | 0.99 |
| Effect of APOE4 Carriage on Change in Plasma Variable Relative to E3/E3 | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Plasma Variable | Reference Diet HSFA/HGI | HM/HGI Diet | HM/LGI Diet | LF/HGI Diet | LF/LGI Diet | Global p | ||||||||||
| Effect | SE | p | Effect | SE | p | Effect | SE | p | Effect | SE | p | Effect | SE | p | ||
| Change in TC | −0.1 | 0.17 | 0.53 | 0.3 | 0.14 | 0.03 | 0.24 | 0.13 | 0.07 | 0.02 | 0.14 | 0.89 | −0.28 | 0.13 | 0.03 | 0.02 |
| Change in LDL-C | −0.10 | 0.14 | 0.49 | 0.23 | 0.12 | 0.05 | 0.05 | 0.11 | 0.65 | 0.07 | 0.12 | 0.57 | −0.23 | 0.11 | 0.04 | 0.07 |
| Change in HDL-C | 0.01 | 0.05 | 0.80 | −0.01 | 0.05 | 0.81 | 0.08 | 0.04 | 0.09 | −0.06 | 0.05 | 0.22 | −0.03 | 0.04 | 0.45 | 0.27 |
| Change in TAG | −0.06 | 0.16 | 0.69 | 0.16 | 0.14 | 0.25 | 0.33 | 0.13 | 0.01 | 0.03 | 0.14 | 0.84 | −0.03 | 0.13 | 0.80 | 0.23 |
| Change in TC: HDL-C ratio | −0.1 | 0.15 | 0.52 | 0.30 | 0.14 | 0.03 | −0.02 | 0.13 | 0.90 | 0.27 | 0.13 | 0.04 | −0.15 | 0.13 | 0.23 | 0.05 |
| Change in Apo B (g/L) | −0.06 | 0.05 | 0.23 | 0.04 | 0.05 | 0.32 | 0.11 | 0.04 | 0.01 | −0.01 | 0.05 | 0.75 | −0.10 | 0.04 | 0.02 | 0.006 |
| Change in Apo A-I (g/L) | 0 | 0.05 | 0.95 | 0.05 | 0.05 | 0.29 | 0.10 | 0.04 | 0.02 | −0.04 | 0.05 | 0.35 | −0.01 | 0.04 | 0.89 | 0.17 |
| Change in ApoB:ApoA-I ratio | −0.06 | 0.04 | 0.13 | −0.01 | 0.03 | 0.84 | 0.04 | 0.03 | 0.17 | 0.02 | 0.03 | 0.43 | −0.07 | 0.03 | 0.02 | 0.05 |
| Effect of APOE2 Carriage on Change in Plasma Variable Relative to E3/E3 | ||||||||||||||||
| Effect | SE | p | Effect | SE | p | Effect | SE | p | Effect | SE | p | Effect | SE | p | ||
| Change in TC | −0.04 | 0.19 | 0.85 | 0.08 | 0.17 | 0.61 | 0.13 | 0.18 | 0.47 | −0.06 | 0.15 | 0.70 | 0.18 | 0.19 | 0.34 | 0.84 |
| Change in LDL-C | 0 | 0.17 | 0.99 | 0.04 | 0.15 | 0.80 | −0.02 | 0.16 | 0.91 | −0.05 | 0.14 | 0.74 | 0.06 | 0.17 | 0.75 | 0.99 |
| Change in HDL-C | 0.03 | 0.06 | 0.67 | 0.03 | 0.05 | 0.57 | −0.04 | 0.06 | 0.51 | −0.01 | 0.05 | 0.88 | 0 | 0.06 | 0.98 | 0.92 |
| Change in TAG | −0.19 | 0.04 | 0,19 | −0.05 | 0.13 | 0.71 | 0.46 | 0.14 | 0.001 | 0 | 0.12 | 0.99 | 0.18 | 0.14 | 0.20 | 0.02 |
| Change in TC:HDL-C ratio | 0.01 | 0.25 | 0.80 | −0.03 | 0.16 | 0.82 | 0.26 | 0.18 | 0.14 | −0.01 | 0.15 | 0.97 | 0.12 | 0.18 | 0.50 | 0.70 |
| Change in ApoB (g/L) | 0 | 0.07 | 0.96 | 0.07 | 0.06 | 0.22 | 0.04 | 0.07 | 0.55 | 0.07 | 0.06 | 0.22 | 0.04 | 0.07 | 0.58 | 0.93 |
| Change in ApoA-I (g/L) | −0.03 | 0.06 | 0.65 | 0.05 | 0.05 | 0.33 | 0.02 | 0.06 | 0.72 | 0.05 | 0.05 | 0.24 | 0.05 | 0.06 | 0.34 | 0.80 |
| Change in ApoB:ApoA-I ratio | 0.01 | 0.04 | 0.74 | 0.03 | 0.04 | 0.48 | 0.02 | 0.04 | 0.65 | 0.03 | 0.04 | 0.38 | 0 | 0.05 | 0.92 | 0.99 |
| Effect of APOE4 Carriage on Change in Plasma Variable Relative to E3/E3 | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Plasma Variable | Reference Diet HSFA/HGI | HM/HGI Diet | HM/LGI Diet | LF/HGI Diet | LF/LGI Diet | Global p | ||||||||||
| Effect | SE | p | Effect | SE | p | Effect | SE | p | Effect | SE | p | Effect | SE | p | ||
| Change in Si | −0.31 | 0.05 | 0.51 | 0.36 | 0.42 | 0.86 | −0.26 | 0.47 | 0.58 | 0.10 | 0.38 | 0.80 | −0.31 | 0.52 | 0.55 | 0.76 |
| Change in AIRg | 154 | 67 | 0.02 | 15 | 60 | 0.80 | 102 | 67 | 0.13 | −33 | 58 | 0.57 | −55 | 73 | 0.45 | 0.13 |
| Change in DI | 521 | 248 | 0.04 | −157 | 224 | 0.48 | 294 | 252 | 0.24 | 9 | 217 | 0.97 | −457 | 274 | 0.10 | 0.06 |
| Change in FPG | −0.11 | 0.16 | 0.50 | −0.06 | 0.14 | 0.66 | 0.04 | 0.16 | 0.80 | 0.23 | 0.13 | 0.09 | −0.04 | 0.17 | 0.82 | 0.50 |
| Change in FPI | 28.0 | 15.4 | 0.07 | −13.8 | 13.8 | 0.32 | 21.2 | 15.2 | 0.17 | 19.2 | 12.7 | 0.13 | −4.1 | 15.6 | 0.80 | 0.18 |
| Effect of APOE2 Carriage on Change in Plasma Variable Relative to E3/E3 | ||||||||||||||||
| Effect | SE | p | Effect | SE | p | Effect | SE | p | Effect | SE | p | Effect | SE | p | ||
| Change in Si | −0.01 | 0.41 | 0.97 | −0.24 | 0.36 | 0.59 | 0.20 | 0.36 | 0.59 | 0.18 | 0.36 | 0.61 | 0.55 | 0.35 | 0.11 | 0.61 |
| Change in AIRg | 97 | 54 | 0.07 | 109 | 47 | 0.02 | 18.9 | 47 | 0.69 | 5 | 47 | 0.92 | −30 | 45 | 0.51 | 0.17 |
| Change in DI | 505 | 199 | 0.01 | 14 | 173 | 0.93 | 112 | 174 | 0.52 | 67 | 174 | 0.70 | 181 | 166 | 0.28 | 0.40 |
| Change in FPG | −0.09 | 0.14 | 0.29 | −0.3 | 0.13 | 0.02 | 0.05 | 0.12 | 0.70 | −0.05 | 0.13 | 0.71 | −0.07 | 0.12 | 0.55 | 0.40 |
| Change in FPI | −12.6 | 28.3 | 0.66 | 8.3 | 24.7 | 0.74 | 57.4 | 22.6 | 0.01 | −2.52 | 24.2 | 0.92 | 0.1 | 23.0 | 0.99 | 0.25 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Griffin, B.A.; Walker, C.G.; Jebb, S.A.; Moore, C.; Frost, G.S.; Goff, L.; Sanders, T.A.B.; Lewis, F.; Griffin, M.; Gitau, R.; et al. APOE4 Genotype Exerts Greater Benefit in Lowering Plasma Cholesterol and Apolipoprotein B than Wild Type (E3/E3), after Replacement of Dietary Saturated Fats with Low Glycaemic Index Carbohydrates. Nutrients 2018, 10, 1524. https://doi.org/10.3390/nu10101524
Griffin BA, Walker CG, Jebb SA, Moore C, Frost GS, Goff L, Sanders TAB, Lewis F, Griffin M, Gitau R, et al. APOE4 Genotype Exerts Greater Benefit in Lowering Plasma Cholesterol and Apolipoprotein B than Wild Type (E3/E3), after Replacement of Dietary Saturated Fats with Low Glycaemic Index Carbohydrates. Nutrients. 2018; 10(10):1524. https://doi.org/10.3390/nu10101524
Chicago/Turabian StyleGriffin, Bruce A., Celia G. Walker, Susan A. Jebb, Carmel Moore, Gary S. Frost, Louise Goff, Tom A. B. Sanders, Fiona Lewis, Margaret Griffin, Rachel Gitau, and et al. 2018. "APOE4 Genotype Exerts Greater Benefit in Lowering Plasma Cholesterol and Apolipoprotein B than Wild Type (E3/E3), after Replacement of Dietary Saturated Fats with Low Glycaemic Index Carbohydrates" Nutrients 10, no. 10: 1524. https://doi.org/10.3390/nu10101524