Effect of Trehalose Supplementation on Autophagy and Cystogenesis in a Mouse Model of Polycystic Kidney Disease


Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Experimental Design
2.3. Histomorphometric Analysis
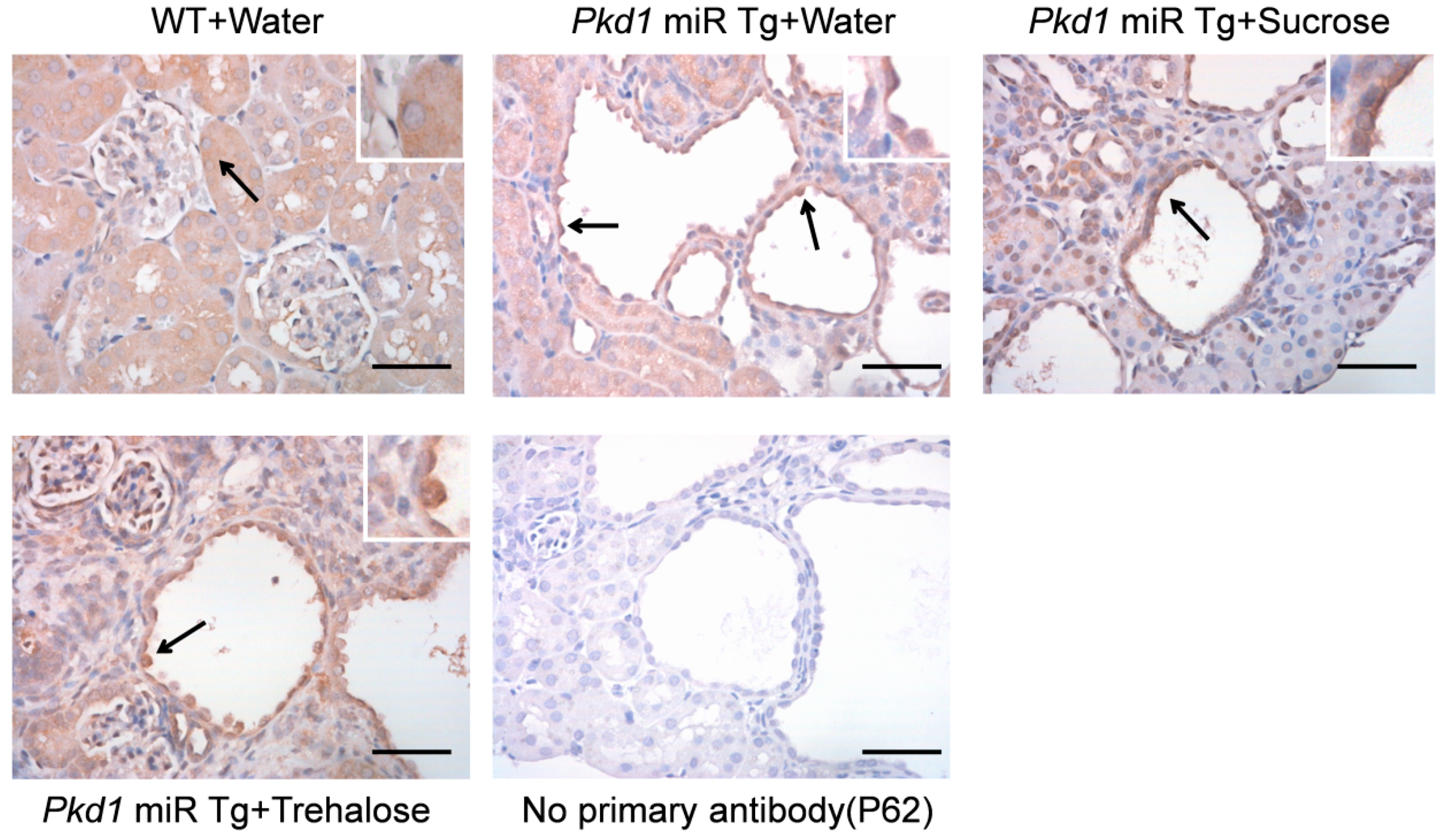
2.4. Immunohistochemistry
2.5. Real-Time Polymerase Chain Reaction
2.6. Western Blotting
2.7. Statistical Analysis
3. Results
3.1. Effect of Trehalose Treatment on Kidney and Body Weight
3.2. Effect of Trehalose Treatment on Renal Function and Blood Sugar
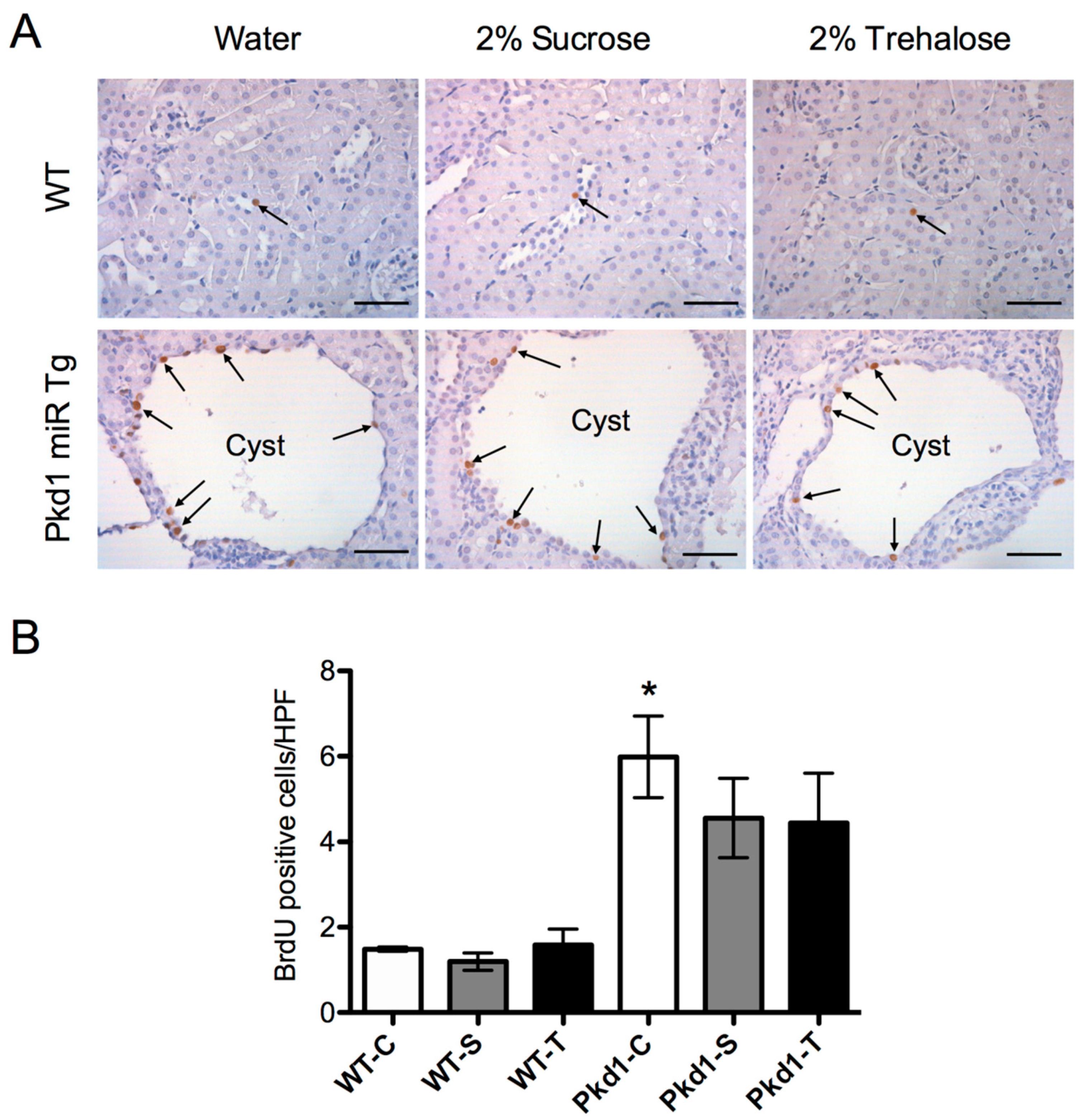
3.3. Effect of Trehalose on Renal Cystogenesis
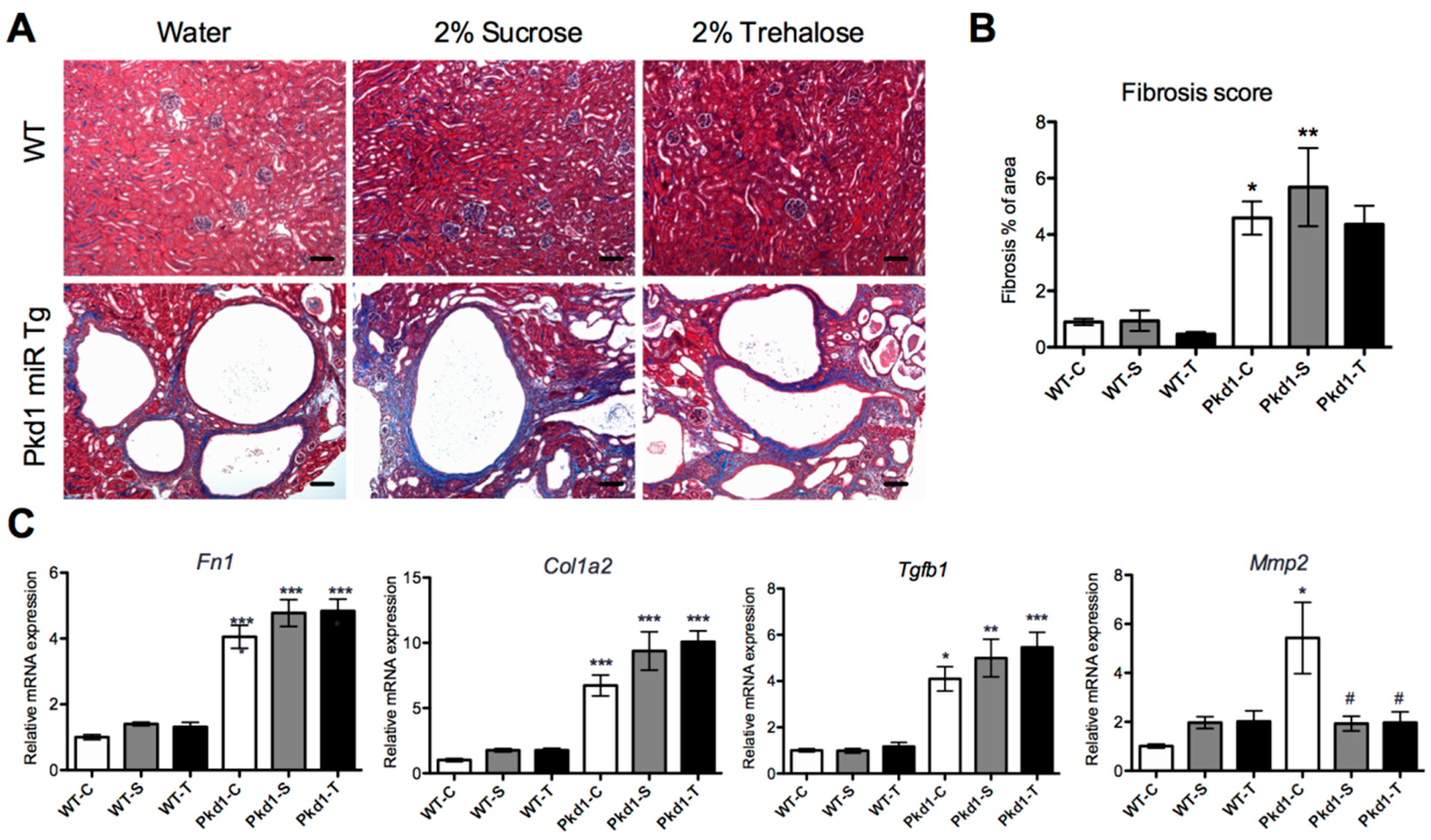
3.4. Effect of Trehalose on Renal Fibrosis
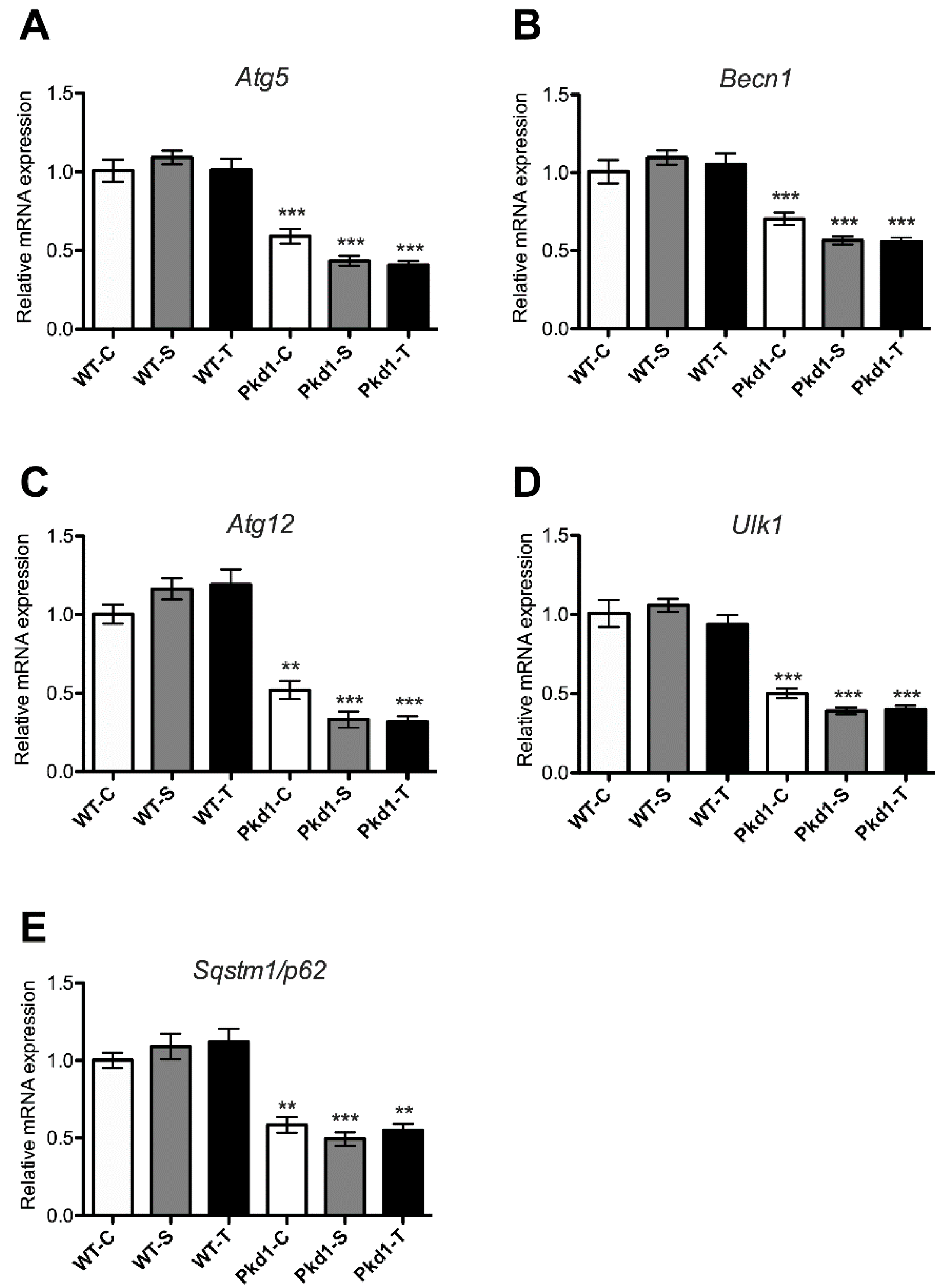
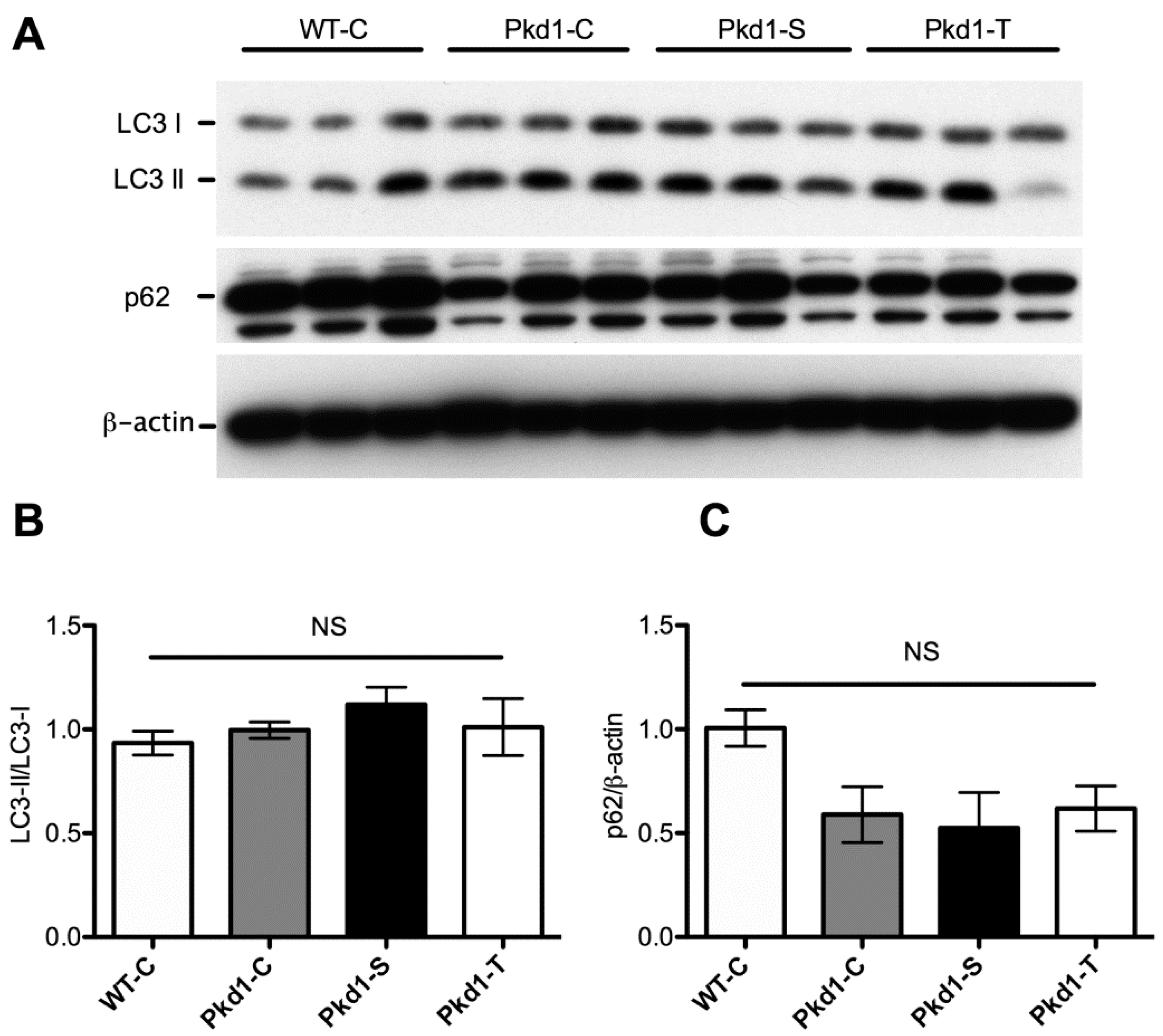
3.5. Effect of Trehalose on Autophagy Activation
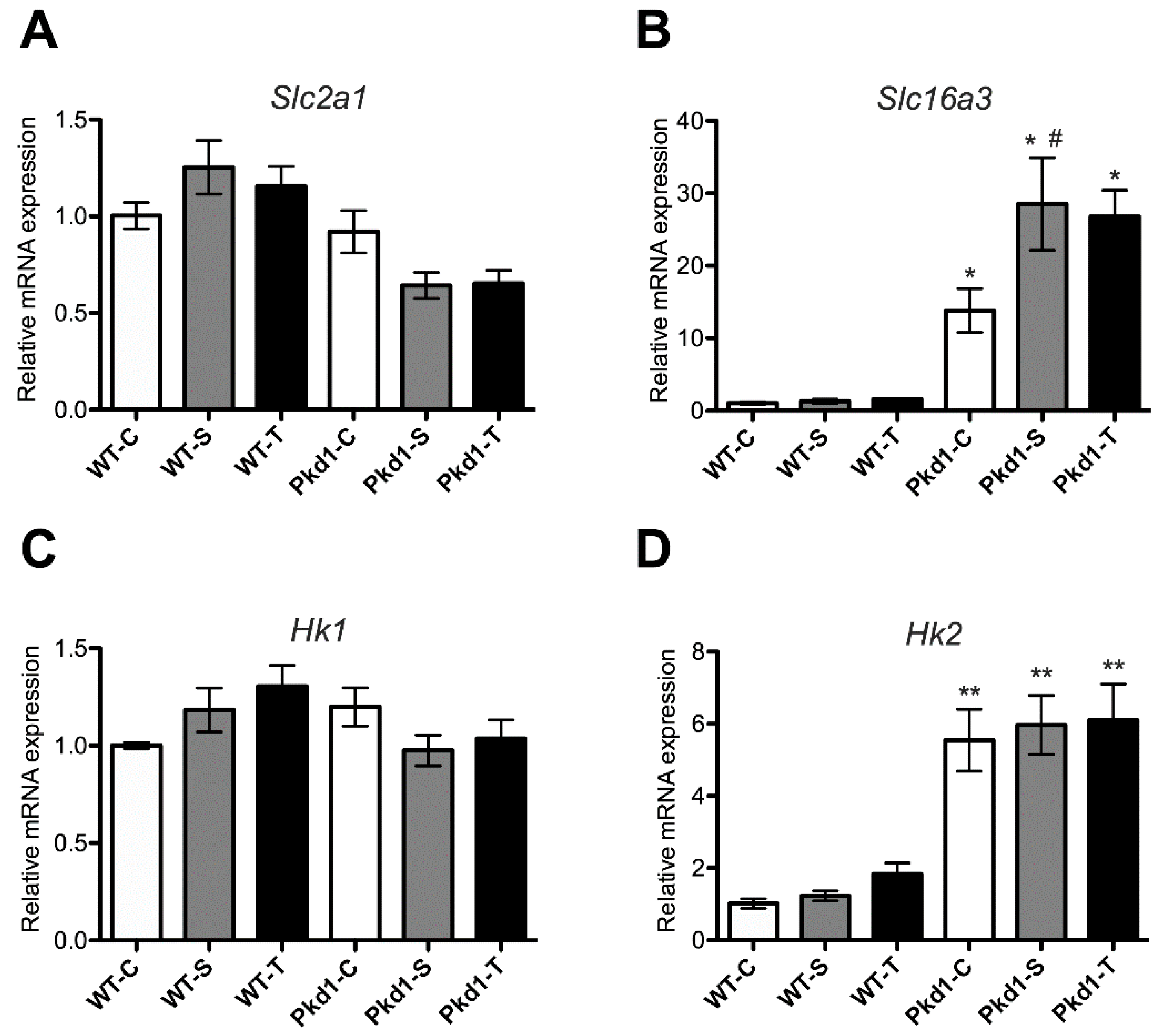
3.6. Effect of Trehalose on Glycolysis
4. Discussion
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Paul, B.M.; Vanden Heuvel, G.B. Kidney: Polycystic kidney disease. Wiley. Interdiscip. Rev. Dev. Biol. 2014, 3, 465–487. [Google Scholar] [CrossRef] [PubMed]
- Harris, P.C.; Rossetti, S. Molecular diagnostics for autosomal dominant polycystic kidney disease. Nat. Rev. Nephrol. 2010, 6, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapin, H.C.; Caplan, M.J. The cell biology of polycystic kidney disease. J. Cell. Biol. 2010, 191, 701–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanktree, M.B.; Chapman, A.B. New treatment paradigms for ADPKD: Moving towards precision medicine. Nat. Rev. Nephrol. 2017, 13, 750–768. [Google Scholar] [CrossRef] [PubMed]
- Torres, V.E.; Harris, P.C. Polycystic kidney disease: Genes, proteins, animal models, disease mechanisms and therapeutic opportunities. J. Intern. Med. 2007, 261, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Aguiari, G.; Catizone, L.; Del Senno, L. Multidrug therapy for polycystic kidney disease: A review and perspective. Am. J. Nephrol. 2013, 37, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Shillingford, J.M.; Murcia, N.S.; Larson, C.H.; Low, S.H.; Hedgepeth, R.; Brown, N.; Flask, C.A.; Novick, A.C.; Goldfarb, D.A.; Kramer-Zucker, A.; et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5466–5471. [Google Scholar] [CrossRef] [Green Version]
- Chang, M.Y.; Ong, A.C.M. Targeting new cellular disease pathways in autosomal dominant polycystic kidney disease. Nephrol. Dial. Transplant. 2018, 33, 1310–1316. [Google Scholar] [CrossRef]
- Ingelfinger, J.R. Tolvaptan and Autosomal Dominant Polycystic Kidney Disease. N. Engl. J. Med. 2017, 377, 1988–1989. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Takiar, V.; Nishio, S.; Seo-Mayer, P.; King, J.D., Jr.; Li, H.; Zhang, L.; Karihaloo, A.; Hallows, K.R.; Somlo, S.; Caplan, M.J. Activating AMP-activated protein kinase (AMPK) slows renal cystogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 2462–2467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belibi, F.; Zafar, I.; Ravichandran, K.; Segvic, A.B.; Jani, A.; Ljubanovic, D.G.; Edelstein, C.L. Hypoxia-inducible factor-1alpha (HIF-1alpha) and autophagy in polycystic kidney disease (PKD). Am. J. Physiol. Ren. Physiol. 2011, 300, F1235–F1243. [Google Scholar] [CrossRef] [PubMed]
- Takakura, A.; Nelson, E.A.; Haque, N.; Humphreys, B.D.; Zandi-Nejad, K.; Frank, D.A.; Zhou, J. Pyrimethamine inhibits adult polycystic kidney disease by modulating STAT signaling pathways. Hum. Mol. Genet. 2011, 20, 4143–4154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, T.B.; Edelstein, C.L.; Hartleben, B.; Inoki, K.; Jiang, M.; Koya, D.; Kume, S.; Lieberthal, W.; Pallet, N.; Quiroga, A.; et al. Emerging role of autophagy in kidney function, diseases and aging. Autophagy 2012, 8, 1009–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, P.; Sieben, C.J.; Xu, X.; Harris, P.C.; Lin, X. Autophagy activators suppress cystogenesis in an autosomal dominant polycystic kidney disease model. Hum. Mol. Genet. 2017, 26, 158–172. [Google Scholar] [CrossRef] [PubMed]
- Tournu, H.; Fiori, A.; Van Dijck, P. Relevance of trehalose in pathogenicity: Some general rules, yet many exceptions. PLoS Pathog. 2013, 9, e1003447. [Google Scholar] [CrossRef] [PubMed]
- Collins, J.; Robinson, C.; Danhof, H.; Knetsch, C.W.; van Leeuwen, H.C.; Lawley, T.D.; Auchtung, J.M.; Britton, R.A. Dietary trehalose enhances virulence of epidemic Clostridium difficile. Nature 2018, 553, 291–294. [Google Scholar] [CrossRef]
- Elbein, A.D.; Pan, Y.T.; Pastuszak, I.; Carroll, D. New insights on trehalose: A multifunctional molecule. Glycobiology 2003, 13, 17R–27R. [Google Scholar] [CrossRef]
- Chen, Q.; Haddad, G.G. Role of trehalose phosphate synthase and trehalose during hypoxia: From flies to mammals. J. Exp. Biol. 2004, 207, 3125–3129. [Google Scholar] [CrossRef]
- Kaplon, R.E.; Hill, S.D.; Bispham, N.Z.; Santos-Parker, J.R.; Nowlan, M.J.; Snyder, L.L.; Chonchol, M.; LaRocca, T.J.; McQueen, M.B.; Seals, D.R. Oral trehalose supplementation improves resistance artery endothelial function in healthy middle-aged and older adults. Aging 2016, 8, 1167–1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizunoe, Y.; Sudo, Y.; Okita, N.; Hiraoka, H.; Mikami, K.; Narahara, T.; Negishi, A.; Yoshida, M.; Higashibata, R.; Watanabe, S.; et al. Involvement of lysosomal dysfunction in autophagosome accumulation and early pathologies in adipose tissue of obese mice. Autophagy 2017, 13, 642–653. [Google Scholar] [CrossRef] [PubMed]
- Emanuele, E. Can trehalose prevent neurodegeneration? Insights from experimental studies. Curr. Drug Targets 2014, 15, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Davies, J.E.; Huang, Z.; Tunnacliffe, A.; Rubinsztein, D.C. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J. Biol. Chem. 2007, 282, 5641–5652. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Machida, Y.; Niu, S.; Ikeda, T.; Jana, N.R.; Doi, H.; Kurosawa, M.; Nekooki, M.; Nukina, N. Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat. Med. 2004, 10, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.L.; Saleem, M.A.; Chan, K.W.; Yung, B.Y.; Law, H.K. Trehalose, an mTOR independent autophagy inducer, alleviates human podocyte injury after puromycin aminonucleoside treatment. PLoS ONE 2014, 9, e113520. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Y.; Yang, H.; Wang, M.G.; Yang, D.B.; Wang, Z.Y.; Wang, L. Trehalose protects against cadmium-induced cytotoxicity in primary rat proximal tubular cells via inhibiting apoptosis and restoring autophagic flux. Cell Death Dis. 2017, 8, e3099. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Hsieh-Li, H.M.; Chiou, Y.Y.; Chien, Y.L.; Ho, H.H.; Chin, H.J.; Wang, C.K.; Liang, S.C.; Jiang, S.T. Progressive renal distortion by multiple cysts in transgenic mice expressing artificial microRNAs against Pkd1. J. Pathol. 2010, 222, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, V.; Lavenir, I.; Ozcelik, S.; Tolnay, M.; Winkler, D.T.; Goedert, M. Stimulation of autophagy reduces neurodegeneration in a mouse model of human tauopathy. Brain 2012, 135, 2169–2177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kara, N.Z.; Toker, L.; Agam, G.; Anderson, G.W.; Belmaker, R.H.; Einat, H. Trehalose induced antidepressant-like effects and autophagy enhancement in mice. Psychopharmacology 2013, 229, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Warner, G.; Hein, K.Z.; Nin, V.; Edwards, M.; Chini, C.C.; Hopp, K.; Harris, P.C.; Torres, V.E.; Chini, E.N. Food Restriction Ameliorates the Development of Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2016, 27, 1437–1447. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Harris, P.C.; Somlo, S.; Batlle, D.; Torres, V.E. Effect of calcium-sensing receptor activation in models of autosomal recessive or dominant polycystic kidney disease. Nephrol. Dial. Transplant. 2009, 24, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Novalic, Z.; van der Wal, A.M.; Leonhard, W.N.; Koehl, G.; Breuning, M.H.; Geissler, E.K.; de Heer, E.; Peters, D.J. Dose-dependent effects of sirolimus on mTOR signaling and polycystic kidney disease. J. Am. Soc. Nephrol. 2012, 23, 842–853. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.Y.; Parker, E.; El Nahas, M.; Haylor, J.L.; Ong, A.C. Endothelin B receptor blockade accelerates disease progression in a murine model of autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 2007, 18, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Norman, J. Fibrosis and progression of autosomal dominant polycystic kidney disease (ADPKD). Biochim. Biophys. Acta 2011, 1812, 1327–1336. [Google Scholar] [CrossRef] [PubMed]
- DeBosch, B.J.; Heitmeier, M.R.; Mayer, A.L.; Higgins, C.B.; Crowley, J.R.; Kraft, T.E.; Chi, M.; Newberry, E.P.; Chen, Z.; Finck, B.N.; et al. Trehalose inhibits solute carrier 2A (SLC2A) proteins to induce autophagy and prevent hepatic steatosis. Sci. Signal. 2016, 9, ra21. [Google Scholar] [CrossRef] [PubMed]
- Lippai, M.; Low, P. The role of the selective adaptor p62 and ubiquitin-like proteins in autophagy. Biomed. Res. Int. 2014, 2014, 832704. [Google Scholar] [CrossRef]
- Bjorkoy, G.; Lamark, T.; Pankiv, S.; Overvatn, A.; Brech, A.; Johansen, T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009, 452, 181–197. [Google Scholar]
- Rodriguez-Navarro, J.A.; Rodriguez, L.; Casarejos, M.J.; Solano, R.M.; Gomez, A.; Perucho, J.; Cuervo, A.M.; Garcia de Yebenes, J.; Mena, M.A. Trehalose ameliorates dopaminergic and tau pathology in parkin deleted/tau overexpressing mice through autophagy activation. Neurobiol. Dis. 2010, 39, 423–438. [Google Scholar] [CrossRef]
- Tanji, K.; Miki, Y.; Maruyama, A.; Mimura, J.; Matsumiya, T.; Mori, F.; Imaizumi, T.; Itoh, K.; Wakabayashi, K. Trehalose intake induces chaperone molecules along with autophagy in a mouse model of Lewy body disease. Biochem. Biophys. Res. Commun. 2015, 465, 746–752. [Google Scholar] [CrossRef]
- Mirzaie, M.; Karimi, M.; Fallah, H.; Khaksari, M.; Nazari-Robati, M. Downregulation of Matrix Metalloproteinases 2 and 9 is Involved in the Protective Effect of Trehalose on Spinal Cord Injury. Int. J. Mol. Cell. Med. 2018, 7, 8–16. [Google Scholar] [PubMed]
- Lenoir, O.; Tharaux, P.L.; Huber, T.B. Autophagy in kidney disease and aging: Lessons from rodent models. Kidney Int. 2016, 90, 950–964. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Boheler, K.R.; Jiang, L.; Chan, C.W.; Tse, W.W.; Keung, W.; Poon, E.N.; Li, R.A.; Yao, X. Polycystin-2 Plays an Essential Role in Glucose Starvation-Induced Autophagy in Human Embryonic Stem Cell-Derived Cardiomyocytes. Stem Cells 2018, 36, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, A. Polycystic kidney disease: Autophagy boost to treat ADPKD? Nat. Rev. Nephrol. 2017, 13, 134. [Google Scholar] [CrossRef] [PubMed]
- Masyuk, A.I.; Masyuk, T.V.; Lorenzo Pisarello, M.J.; Ding, J.F.; Loarca, L.; Huang, B.Q.; LaRusso, N.F. Cholangiocyte autophagy contributes to hepatic cystogenesis in polycystic liver disease and represents a potential therapeutic target. Hepatology 2018, 67, 1088–1108. [Google Scholar] [CrossRef] [PubMed]
- Rowe, I.; Chiaravalli, M.; Mannella, V.; Ulisse, V.; Quilici, G.; Pema, M.; Song, X.W.; Xu, H.; Mari, S.; Qian, F.; et al. Defective glucose metabolism in polycystic kidney disease identifies a new therapeutic strategy. Nat. Med. 2013, 19, 488–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck Gooz, M.; Maldonado, E.N.; Dang, Y.; Amria, M.Y.; Higashiyama, S.; Abboud, H.E.; Lemasters, J.J.; Bell, P.D. ADAM17 promotes proliferation of collecting duct kidney epithelial cells through ERK activation and increased glycolysis in polycystic kidney disease. Am. J. Physiol. Ren. Physiol. 2014, 307, F551–F559. [Google Scholar] [CrossRef] [PubMed]
- Kraus, A.; Schley, G.; Kunzelmann, K.; Schreiber, R.; Peters, D.J.; Stadler, R.; Eckardt, K.U.; Buchholz, B. Glucose promotes secretion-dependent renal cyst growth. J. Mol. Med. 2016, 94, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Chiaravalli, M.; Rowe, I.; Mannella, V.; Quilici, G.; Canu, T.; Bianchi, V.; Gurgone, A.; Antunes, S.; D’Adamo, P.; Esposito, A.; et al. 2-Deoxy-d-Glucose Ameliorates PKD Progression. J. Am. Soc. Nephrol. 2016, 27, 1958–1969. [Google Scholar] [CrossRef] [PubMed]
- Nagao, S.; Nishii, K.; Katsuyama, M.; Kurahashi, H.; Marunouchi, T.; Takahashi, H.; Wallace, D.P. Increased water intake decreases progression of polycystic kidney disease in the PCK rat. J. Am. Soc. Nephrol. 2006, 17, 2220–2227. [Google Scholar] [CrossRef]
- Chang, M.Y.; Hsieh, C.Y.; Lin, C.Y.; Chen, T.D.; Yang, H.Y.; Chen, K.H.; Hsu, H.H.; Tian, Y.C.; Chen, Y.C.; Hung, C.C.; et al. Effect of celastrol on the progression of polycystic kidney disease in a Pkd1-deficient mouse model. Life Sci. 2018, 212, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Mizunoe, Y.; Kobayashi, M.; Sudo, Y.; Watanabe, S.; Yasukawa, H.; Natori, D.; Hoshino, A.; Negishi, A.; Okita, N.; Komatsu, M.; et al. Trehalose protects against oxidative stress by regulating the Keap1-Nrf2 and autophagy pathways. Redox Biol. 2018, 15, 115–124. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | WT/C (n = 3) | WT/S (n = 6) | WT/T (n = 9) | PKD1/C (n = 9) | PKD1/S (n = 8) | PKD1/T (n = 7) |
|---|---|---|---|---|---|---|
| Body weight (g) | 29.4 ± 1.0 | 28.4 ± 0.4 | 27.2 ± 0.3 | 26.7 ± 1.0 | 24.6 ± 0.6 * | 26.1 ± 0.6 |
| Kidney weight (g) | 0.343 ± 0.025 | 0.319 ± 0.008 | 0.316 ± 0.001 | 0.592 ± 0.019 * | 0.569 ± 0.046 * | 0.584 ± 0.041 * |
| Kidney-to-body weight Ratio (%) | 1.16 ± 0.04 | 1.12 ± 0.04 | 1.16 ± 0.03 | 2.23 ± 0.07 * | 2.30 ± 0.17 * | 2.23 ± 0.13 * |
| Plasma BUN (mg/dL) | 23.2 ± 1.1 | 25.2 ± 1.0 | 27.9 ± 1.1 | 42.1 ± 1.8 * | 45.9 ± 2.7 * | 45.0 ± 3.5 * |
| Plasma cystatin C (ng/mL) | 456.4 ± 11.1 | 454.2 ± 48.1 | 510.0 ± 26.8 | 600.7 ± 39.8 | 686.9 ± 47.5 * | 645.1 ± 35.5 |
| Blood sugar(mg/dL) | 155.0 ± 9.6 | 213.8 ± 16.7 | 216.3 ± 12.4 | 196.8 ± 14.9 | 216.0 ± 12.6 | 215.8 ± 11.2 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chou, L.-F.; Cheng, Y.-L.; Hsieh, C.-Y.; Lin, C.-Y.; Yang, H.-Y.; Chen, Y.-C.; Hung, C.-C.; Tian, Y.-C.; Yang, C.-W.; Chang, M.-Y. Effect of Trehalose Supplementation on Autophagy and Cystogenesis in a Mouse Model of Polycystic Kidney Disease. Nutrients 2019, 11, 42. https://doi.org/10.3390/nu11010042
Chou L-F, Cheng Y-L, Hsieh C-Y, Lin C-Y, Yang H-Y, Chen Y-C, Hung C-C, Tian Y-C, Yang C-W, Chang M-Y. Effect of Trehalose Supplementation on Autophagy and Cystogenesis in a Mouse Model of Polycystic Kidney Disease. Nutrients. 2019; 11(1):42. https://doi.org/10.3390/nu11010042
Chicago/Turabian StyleChou, Li-Fang, Ya-Lien Cheng, Chun-Yih Hsieh, Chan-Yu Lin, Huang-Yu Yang, Yung-Chang Chen, Cheng-Chieh Hung, Ya-Chung Tian, Chih-Wei Yang, and Ming-Yang Chang. 2019. "Effect of Trehalose Supplementation on Autophagy and Cystogenesis in a Mouse Model of Polycystic Kidney Disease" Nutrients 11, no. 1: 42. https://doi.org/10.3390/nu11010042