1. Introduction
Mesenchymal stromal cells (MSCs) constitute a major cell population which is used in research, translational studies, and clinical applications, and bone marrow is one major tissue source. MSCs are largely isolated and described by minimal criteria which were defined by the International Society for Cell Therapy in 2006: Fibroblastic cells with plastic-adherent growth in cell culture, with the presence and absence of selected cell surface antigens and with the capacity to differentiate into osteoblasts, adipocytes, and chondrocytes in vitro [
1,
2]. Unfortunately, cell culture conditions and methods for examination of these criteria vary largely between individual labs. In research studies bone marrow is frequently obtained from patients during diagnostic or therapeutic intervention. The health state of these donors might be unclear. Age, gender, and other parameters such as inflammatory status have an important but largely uncontrollable impact and consequently calls for action to obtain more information. Furthermore it is known that culture conditions including the choice of fetal calf serum and use of different proteases during passaging can have an immense effect on the performance of the cells. To start at the very beginning with tissue harvest for MSC isolation, it might be important to know how the bone marrow is harvested and treated before extraction with respect to anti-coagulation—next to additional interesting aspects like the site (iliac crest or long bones) and mode of bone marrow harvest (aspiration or retrieval from an opened bone).
In the published literature, many publications do not give any information on whether their bone marrow was subjected to anticoagulant treatment in order to prevent clotting. Secondly, if so, the chemical nature of the used anticoagulant is often not indicated. Different anticoagulants are in current use for blood and bone marrow: Acid citrate dextrose, citrate phosphate dextrose, ethylenediamine tetraacetic acid, heparin (Li
+-, Na
+-, K
+-variants), and others. Thirdly, the applied dosages vary largely. Some studies indicate that mere rinsing of syringes with heparin solution was applied [
3] but others used 100 IU/mL bone marrow [
4]. Two types of heparins are widely used, high molecular weight unfractionated heparin and low molecular weight fractionated heparin. In general, there is not only a difference in molecular weight, it is also shown that there is a more unpredictable effect for unfractionated heparin [
5]. Chemically, heparin constitutes a glycosaminoglycan. It acts primarily through a complex with antithrombin III thereby preventing formation of fibrin from fibrinogen and consequently prevents blood clotting. In addition, with a comparably high degree of sulfation and consequently strong negative charge, heparin belongs to a promising class of bio-active substances that can potentially enhance the biological effects of proteins due to binding and sustained and controlled release of cytokines including growth factors (reviewed recently by [
6]).
The issue of anticoagulation may become particularly relevant when systemic administration of MSCs is planned. Therefore, several studies have addressed the effect of heparin on performance of bone marrow MSCs (BM-MSCs). BM-MSCs from different species were applied intravenously in animal models in which heparin co-application successfully prevented cell coagulation in vivo [
7]. Human BM-MSCs injected into infarcted rabbit hearts significantly reduced the fibrotic area when combined with subcutaneous application of heparin [
8]. Non-modified and modified heparin was also used as an appealing biomaterial in many studies in combination with MSCs and other cell types, attractive for local use in vitro and in vivo [
9,
10]. Here, heparin seemed to facilitate and increase the proliferation of MSCs.
Studies have also been performed with constant heparin addition during BM-MSC culture in vitro [
11,
12]. Promotion of osteogenic and reduction of adipogenic differentiation were found upon use of 20 U/mL unfractionated heparin [
11]. Contrasting with these results, another study showed that osteogenic, chondrogenic, and adipogenic differentiation were scarcely affected by long-term culture with 160 ng/mL heparin [
12]. The expression of the surface antigens CD49a, CD73, and CD105 did not show notable changes after passaging in the presence of 160 ng/mL heparin [
12]. Dependent on the concentration of heparin and the MSC passage, different effects on proliferation were observed [
12]. The authors concluded that heparin should not be used as culture supplement for expansion and maintenance of naïve human MSCs [
10].
Effects of heparin addition to bone marrow preparations and the ensuing consequences for isolation and functionality of BM-MSCs have not yet been investigated systematically. One study showed that heparin blocked SDF1/CXCR4-signaling in mononuclear cells from bone marrow (without expansion in adherent cell culture), thereby interfering with homing and migration of the cells [
13]. However, culture-expanded MSCs derived from these mononuclear cells were not investigated in the study [
13]. In another analysis, human BM-MSCs as well as stromal cells from umbilical cord and white adipose tissue were isolated and propagated in human platelet lysate-supplemented media with or without heparin [
14]. Here, transcriptome analyses revealed regulation of distinct gene sets by heparin including signaling cascades involved in proliferation, cell adhesion, apoptosis, inflammation, and angiogenesis, depending on the stromal cell origin [
14]. Despite these observations heparin had no substantial effect on long-term proliferation and in vitro tri-lineage differentiation of stromal cells [
14].
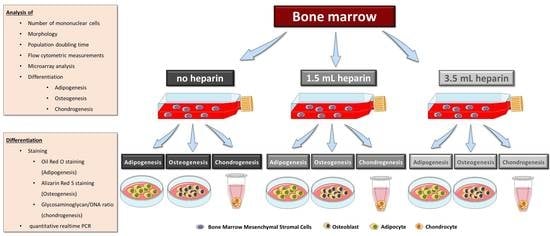
Considering this state of knowledge, we were interested in obtaining more detailed information on the influence of heparin on human bone marrow during the initial step for subsequent MSC isolation—the tissue sampling process itself—and the quality of the subsequently isolated MSCs. This was reinforced by our finding that, despite the use of standard protocols for cell isolation and differentiation assays, the large majority of our cell preparations obtained from different human orthopedic bone marrow donors did not conform well with the International Society for Cell Therapy minimal criteria, in particular with respect to their in vitro differentiation potential into osteogenic or chondrogenic lineages. Colleagues from our university reported similar findings for bone marrow samples from healthy volunteers (unpublished results). Therefore, the present study was designed with twelve bone marrow donations obtained during implantation of a total hip arthroplasty. Three aliquots of bone marrow from the same donor, 5 mL each, were either not anti-coagulated or supplemented with two different concentrations of heparin. Mononuclear cells including MSCs were subsequently isolated by density gradient centrifugation within four hours of bone marrow harvest. Plastic-adherent cells were expanded and characterized for the number of mononuclear cells obtained after density gradient centrifugation, morphology, proliferation, expression of cell surface antigens, in vitro differentiation, and gene expressions.
2. Materials and Methods
2.1. Materials
Heparin was supplied as sodium salt, from pig intestine mucosa, unfractionated (UFH), distributor Rotexmedica (25.000 IU/ 5 mL).
2.2. Ethics Statement
Ethical approval was obtained from the ethical committee of Hannover Medical School (#565-2009, 28.08.2009, #565-2016, 22.04.2016). Samples were collected in accordance with the Declaration of Helsinki of 1975, revised in 2013, after written informed consent of all donors. All personal information except age and gender was deleted.
2.3. Bone Marrow Harvest
No preconditioning of the patients was performed before surgery. In particular systemic anticoagulation was not used. Donor information is presented in
Table 1 and
Supplemental Table S1. During preparation of the femoral cavity for the implantation of the femoral stem for a total hip arthroplasty, the femoral canal was opened, bone marrow was pressed out and collected intraoperatively as described [
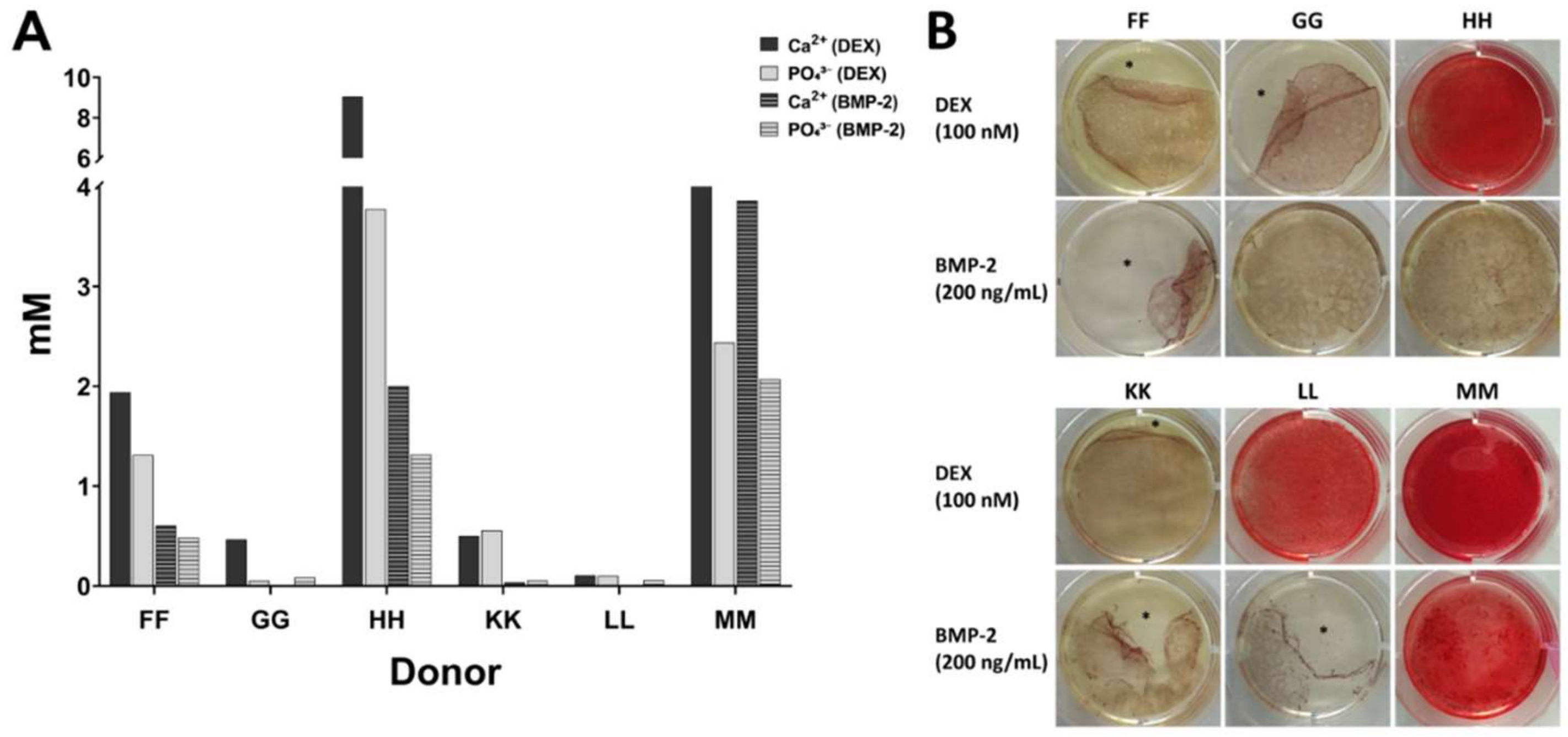
15]. During this process, no coagulation was observed. For each donor, three aliquots of 5 mL bone marrow were collected and each sample was prepared as follows: Condition 1 with no heparin in the syringe, condition 2 supplemented with 1.5 mL heparin (i.e., 1,500 IU/mL bone marrow), and condition 3 supplemented with 3.5 mL heparin (i.e., 3500 IU/mL bone marrow). The quality of bone marrow was assessed intraoperatively macroscopically and graded from 1 (excellent) to 6 (inferior) (
Table 2). “Excellent” means that the bone marrow fluid contained a high amount of bone marrow cells and only few blood cells, i.e., was not reddish but yellow turbid due to the presence of fat cells. “Inferior” designates a small percentage of bone marrow cells and a high percentage of blood. In addition, the sequence of sample preparations was varied intraoperatively i.e., the sample without heparin was not always the first sample to be prepared (
Table 2) in order to avoid introducing bias. The bone marrow samples were processed in the laboratory for isolation of mononuclear cells by density gradient centrifugation as quickly as possible, usually within 4 h. Only bone marrow from donor J was stored overnight. For processing of identical volumes, 0.9% sodium chloride (B. Braun, Melsungen, Germany) were used to make up a final sample volume of 8.5 mL: the sample with no heparin was diluted with 3.5 mL 0.9% NaCl, the sample with 1.5 mL heparin was diluted with 2 mL 0.9% NaCl and the sample with 3.5 mL heparin was not diluted. Sodium chloride was chosen as it has similarities to the commercial heparin preparation which contained benzyl alcohol, sodium chloride, and water for injection (exact composition proprietary knowledge of the manufacturer).
2.4. Isolation and Culture of Human MSCs from Bone Marrow
The bone marrow preparations were further diluted fourfold with phosphate-buffered saline without Ca
2+, Mg
2+ (PBS). The samples were filtrated (cell strainer, 100 µm) to remove bone debris and then were slowly applied to the density gradient material using a ratio of 1 volume Biocoll (Biochrom, Berlin, Germany, L6115; ρ = 1.077 g/mL) to 2 volumes of diluted bone marrow in conical 50 mL centrifuge tubes. After centrifugation at 500×
g for 30 min at room temperature without brakes, the mononuclear cells directly above the density gradient material were recovered, washed once with PBS, pelleted and suspended in 10 mL medium. Cell counting was performed manually with a Fuchs-Rosenthal chamber with the cell suspensions diluted 1:10. The retrieved mononuclear cells were cultured in vessels for growth of adherent cells at a plating density of 500,000 mononuclear cells/cm
2. The MSC expansion medium was Dulbecco’s Modified Eagle´s Medium (DMEM) Low Glucose (1 g/L glucose, Biochrom, FG0415) supplemented with 10% (
v/
v) fetal bovine serum (not heat-inactivated, Thermo Fisher Scientific, Schwerte, Germany, “HyClone”, SV30160.03), 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES, Biochrom), 1% (100 U/mL/100 µg/mL) penicillin/streptomycin (Biochrom) and 2 ng/mL human recombinant fibroblast growth factor-2 (FGF-2, from
Escherichia coli, PeproTech, Hamburg, Germany). The cells were cultured at 37 °C with 5% CO
2 at 85% humidity. After 24 h the medium was replaced by fresh MSC expansion medium and subsequent media changes were performed every 3–4 days. The cells were passaged at a confluence of around 70% by using 0.05%/0.02% (
w/
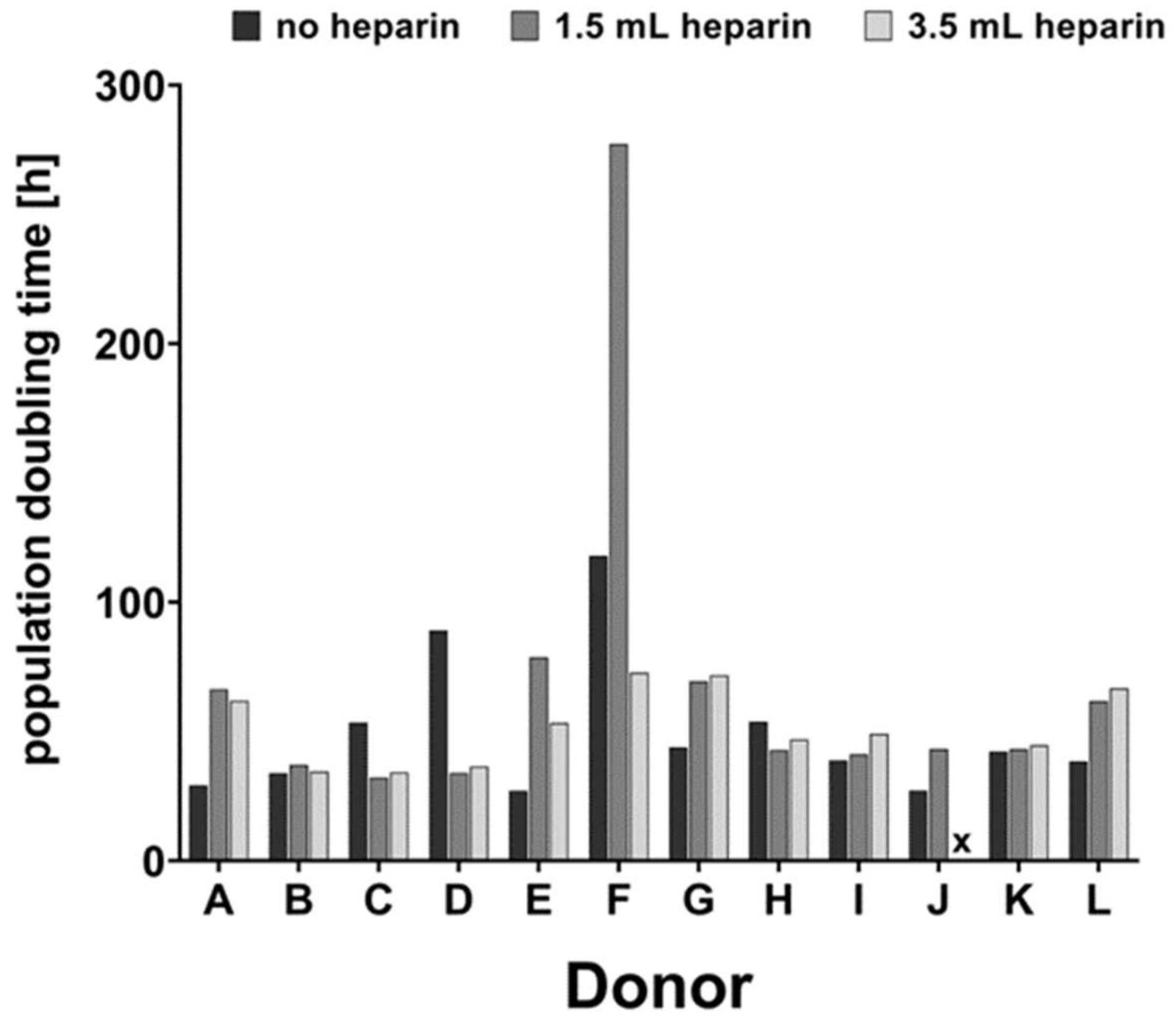
v) trypsin-ethylenediamine tetraacetic acid solution (Trypsin/EDTA; Biochrom L2153) and passaged with 2000 cells per cm² for a maximum of five passages. Morphology was documented by phase contrast microscopy. Proliferation was documented by manual cell-counting with a Fuchs-Rosenthal chamber. Population doubling times (PDT, in hours) were calculated between passages 1 and 2 using the formula:
Detailed information on isolation and propagation of the different MSC preparations is compiled in
Table 2. Bone marrow from twelve donors (A to L) was compared in the present study. In addition, preliminary experiments were conducted with cell samples from donors AA—SS which had been obtained previously by our regular procedure of mixing 2.5 mL heparin with 10 mL bone marrow (1.250 IU/mL bone marrow).
2.5. Flow Cytometric Measurements (FCM)
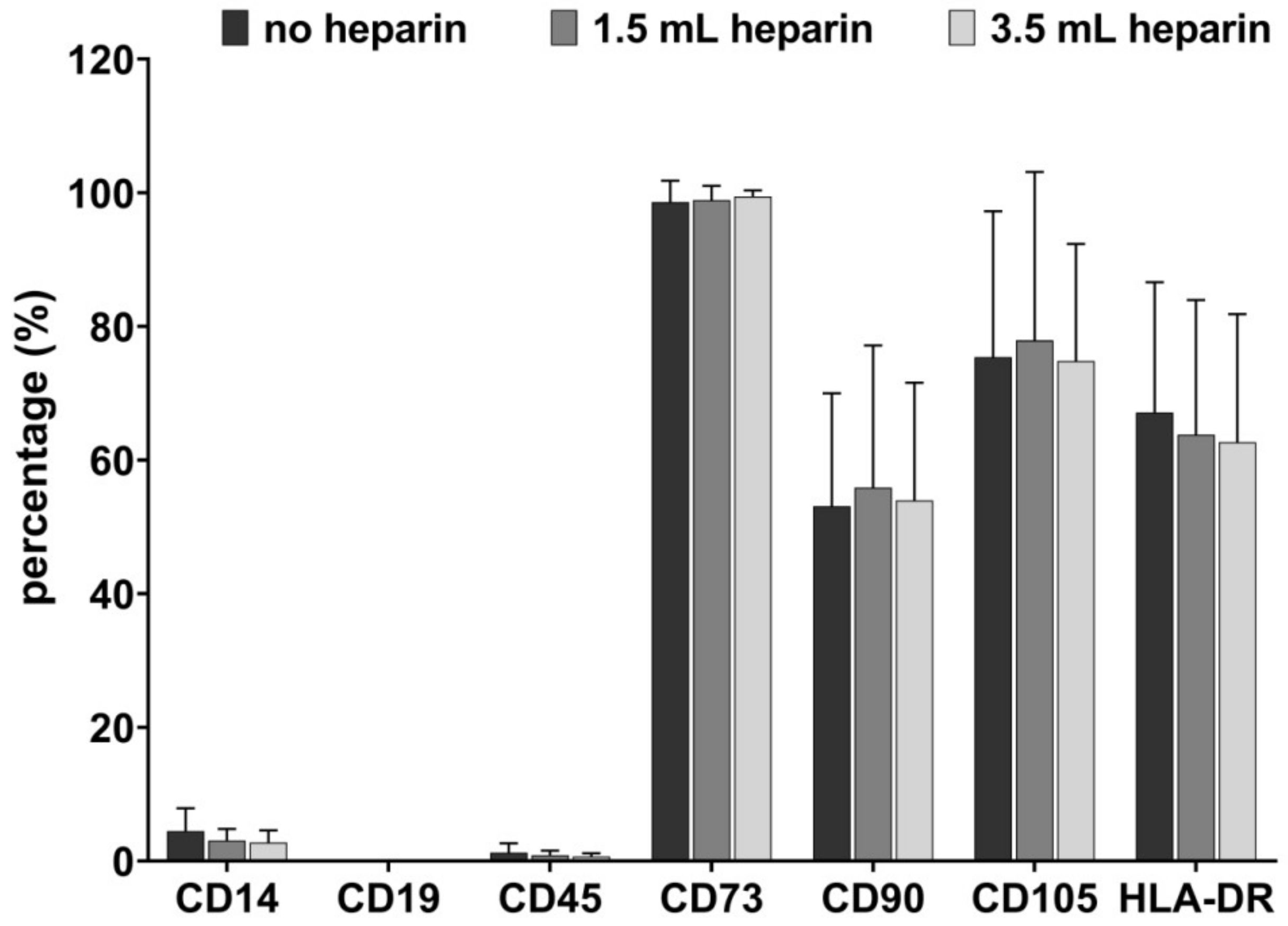
One T25 flask was used per condition and donor after the cells had reached a confluence of about 80% in passages 2 to 4. After washing with PBS, the cells were detached with Trypsin/EDTA and portioned into 5 mL round-bottom test tubes (Corning, Bedford, USA). Blocking was not required due to a specifically mutated human IgG1 Fc region which eliminates their binding to Fc receptors. Samples were subsequently stained with antibodies against CD14, CD19, CD45, CD73, CD90, CD105, HLA-DR, and the respective isotype-matching IgG control antibodies diluted in PBS with 2% (w/v) bovine serum albumin (BSA, 100 µl/ sample) at 4 °C for 15 min. The following monoclonal anti-human antibodies were used: CD14-APC (allophycocyanin, clone REA599, IgG1; Miltenyi Biotech, Bergisch-Gladbach, Germany), CD19-Viogreen (clone REA675, IgG1; Miltenyi Biotech), CD45-APC-Vio770 (clone REA747, IgG1; Miltenyi Biotech), CD73-PEVio615 (clone REA804, IgG1; Miltenyi Biotech), CD90-FITC (fluorescein isothiocyanate; clone REA897, IgG1; Miltenyi Biotech), CD105-Vioblue (clone REA794, IgG1; Miltenyi Biotech), HLA-DR-PE (human leukocyte antigen-DR with phycoerythrin; clone REA805, IgG1; Miltenyi Biotech). Afterwards, cells were washed twice with PBS/BSA and analyzed by flow cytometry using MACSQuant10 (Miltenyi Biotech) and FCSalyzer 0.9.14-alpha software. The entire flow cytometric procedure was performed by one person for all samples to minimize individual variance with respect to gating.
2.6. In Vitro Differentiation of MSCs
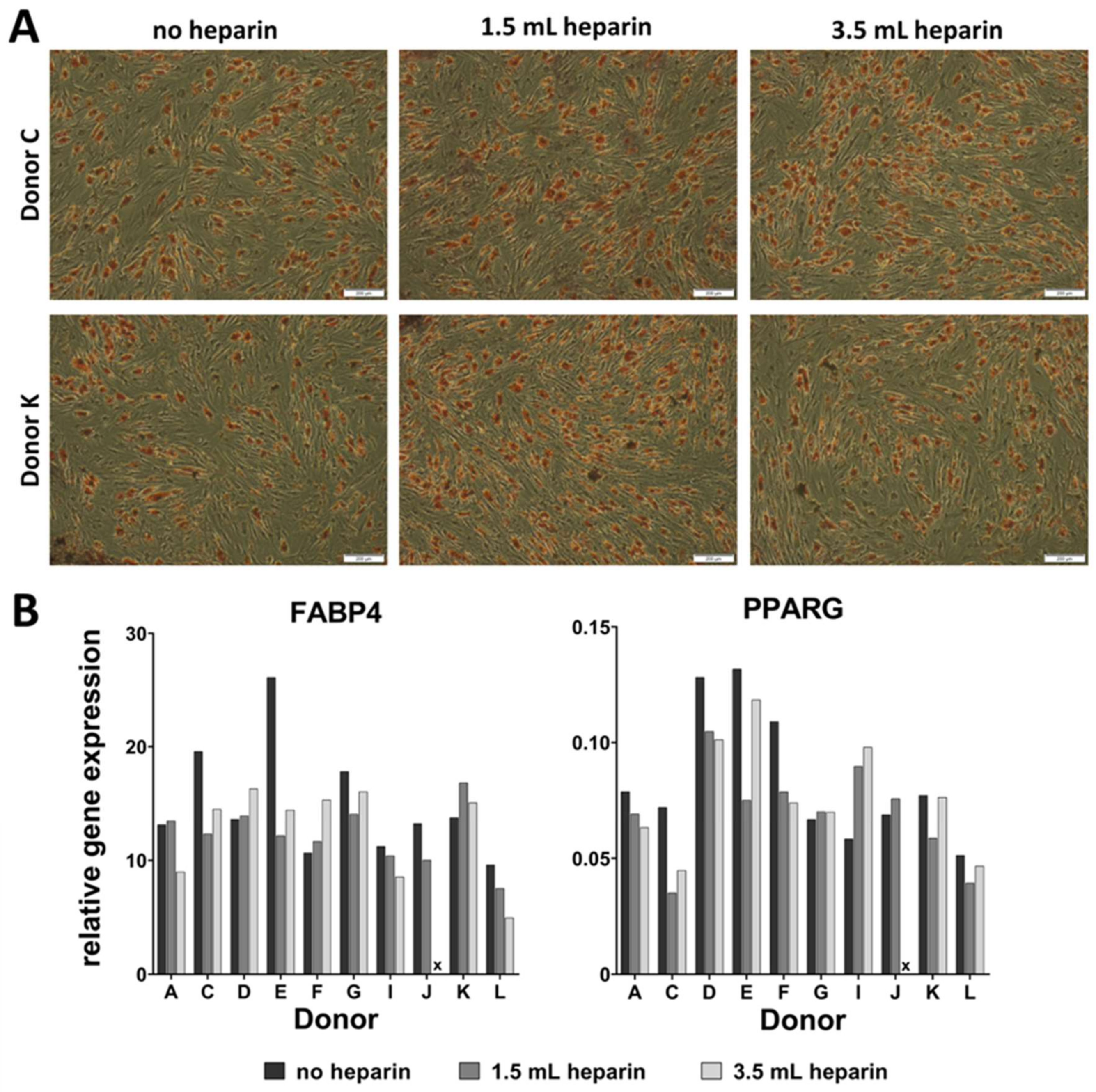
Adipogenic differentiation was induced by plating 5000 cells/cm2 in sixwell-plates (Greiner, Frickenhausen, Germany). Upon reaching confluence, differentiation was induced by using DMEM with high glucose content (4.5 g/L glucose, Biochrom, Berlin, Germany, FG0435) with 10% (v/v) fetal bovine serum, 20 mM HEPES, 1% (100 U/mL/100 µg/mL) penicillin/streptomycin (Biochrom) and supplemented with 1 µM dexamethasone, 60 µM indomethacine (Sigma, Munich, Germany), 500 µM 3-isobutyl-1-methylxanthine (Sigma), and 10 µg/mL insulin (from bovine pancreas, Sigma I0516). Differentiation was performed for a maximum of 14 days and medium was replaced twice a week. RNA was isolated from all samples as described below.
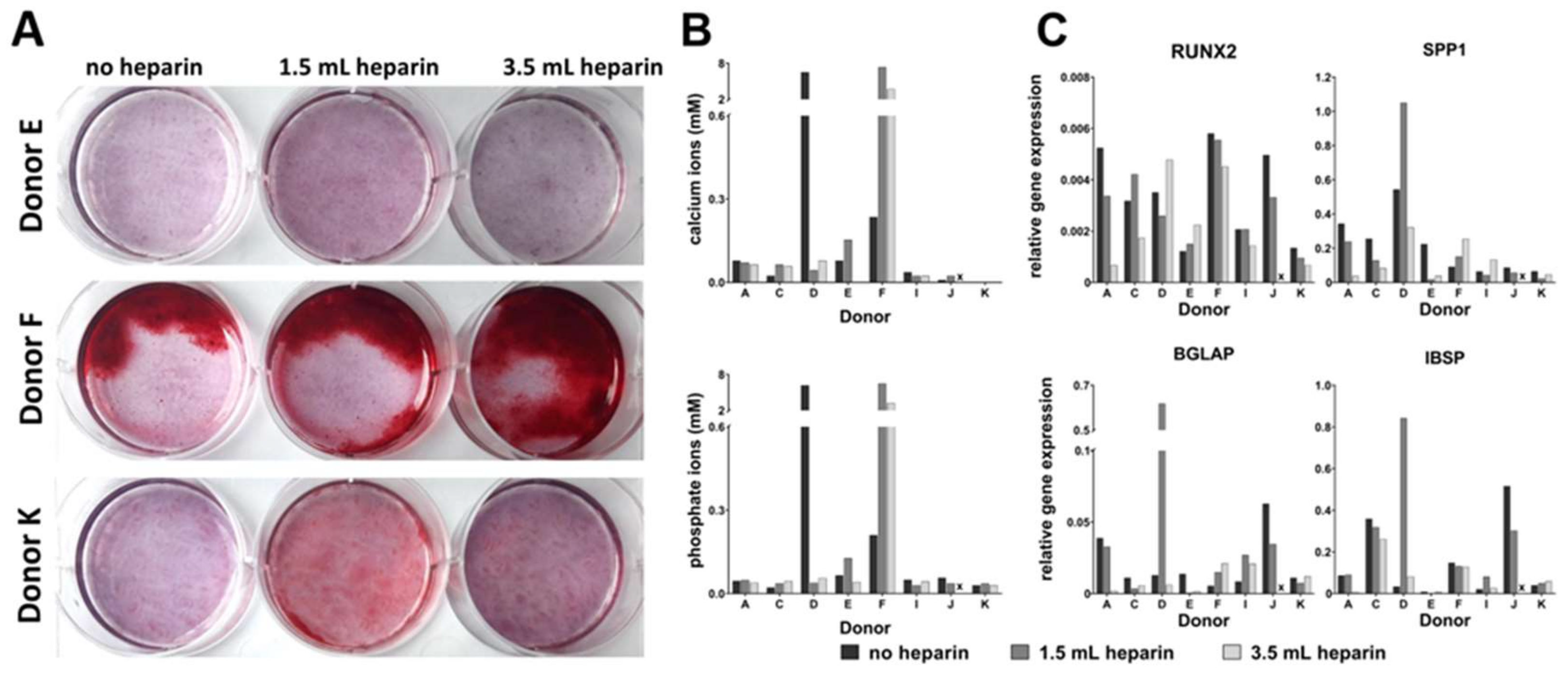
Osteogenic differentiation was induced by plating 5000 cells/cm
2 in sixwell-plates (Greiner) coated with 2% (
w/
v) gelatin B (dissolved in water; Sigma G6650 from bovine skin, autoclaved). Upon reaching confluence (termed day 0 of differentiation), differentiation was induced by supplementation of basal medium consisting of DMEM with Low Glucose with 10% (
v/
v) fetal bovine serum, 20 mM HEPES, 1% (100 U/mL / 100 µg/mL) penicillin/streptomycin (Biochrom) with either 100 nM dexamethasone (Sigma) or 200 ng/mL recombinant human bone morphogenetic protein-2 (BMP-2, from
Escherichia coli, kind gift from G. Gross, Helmholtz Centre for Infection Research, Braunschweig [
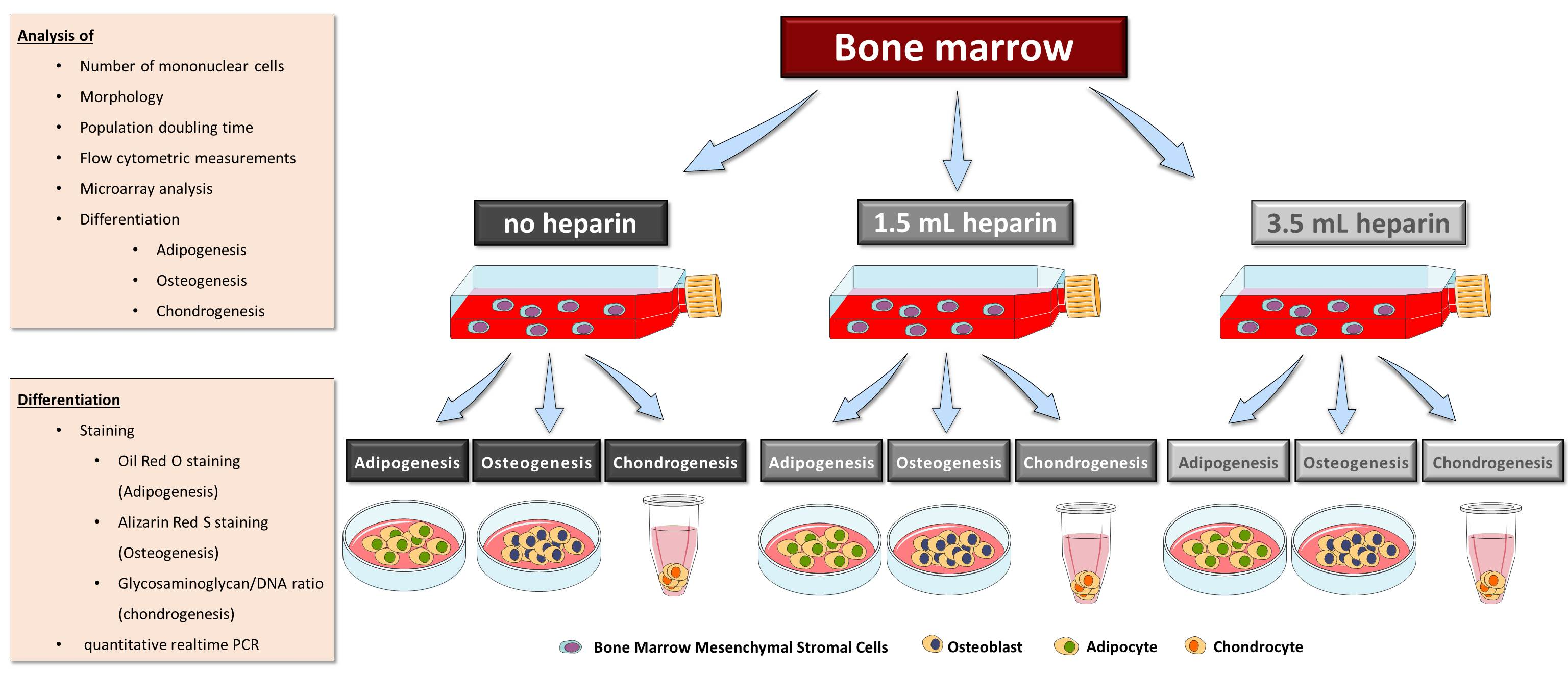
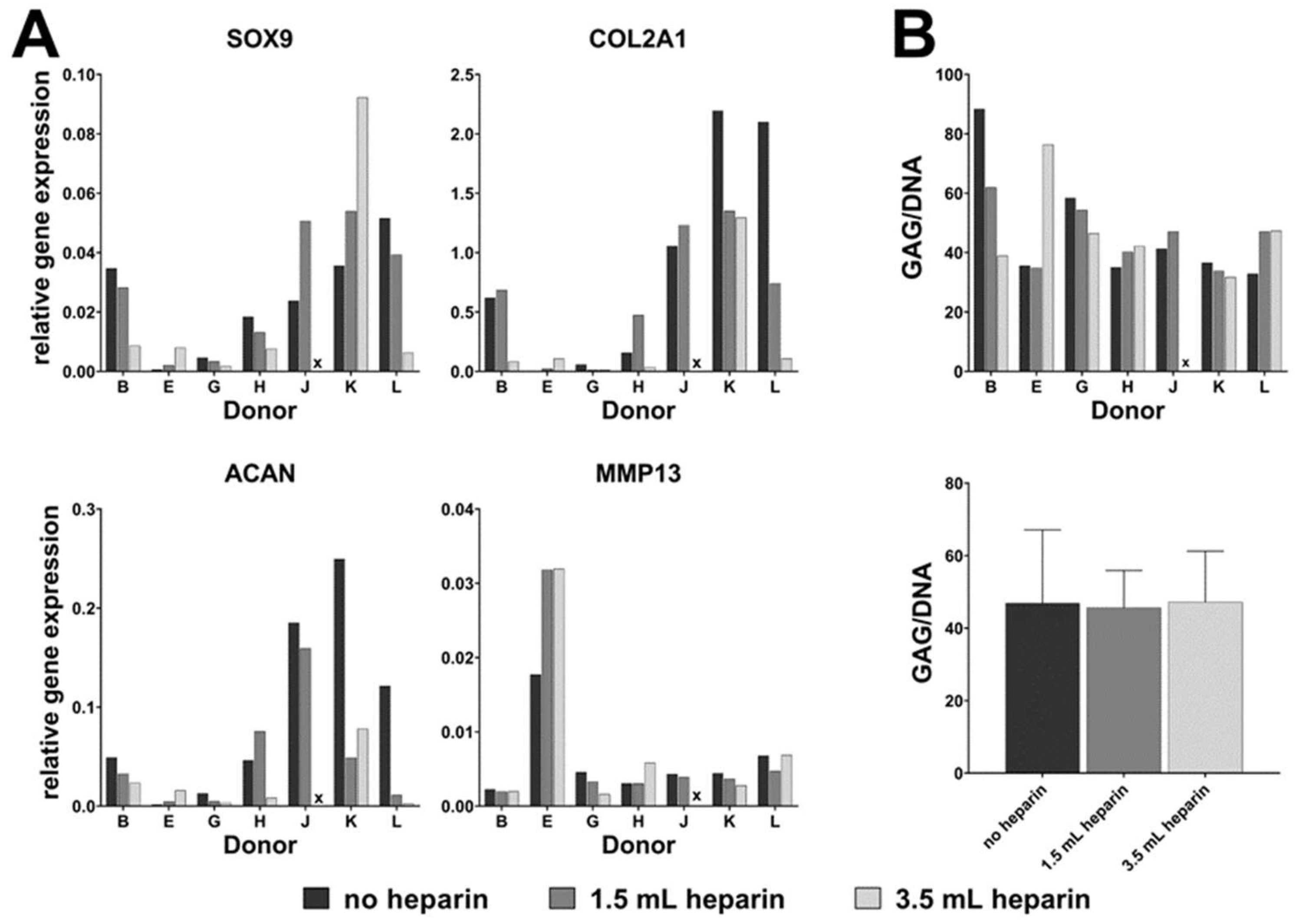
16]) as indicated in the results, plus 50 µM ascorbate-2-phosphate (Sigma) and 10 mM beta-glycerophosphate (Sigma) in both induction protocols. Differentiation was performed for 27 days. Medium was replaced twice a week. MSCs from donor G started to detach at day 19 of differentiation. Therefore, this donor was not included in the analyses. Available cell numbers from donor L were too low so that no osteogenic differentiation experiment was started. RNA was isolated from all samples as described below. Parallel cultures were fixed for cytochemical staining or for determination of the calcium-to-phosphate ratio in the cell layer as described below.
Chondrogenic differentiation was induced in a three-dimensional pellet culture. The required number of cells (for each pellet 1.25 × 105 cells) was transferred into a 15 mL-tube (Greiner) and centrifuged for 5 min at 200× g. The supernatant was removed and the pellet washed with DMEM with high glucose content (Biochrom, FG0435) with 20 mM HEPES, 1% (100 U/mL / 100 µg/mL) penicillin/streptomycin (Biochrom). After another centrifugation step (5 min at 200× g), the medium was discarded and the generated pellet was mixed with induction medium made of DMEM with high glucose content (Biochrom, FG0435) with 20 mM HEPES, 1% (100 U/mL / 100 µg/mL) penicillin/streptomycin (Biochrom), 100 nM dexamethasone, 1x ITS+ (Sigma I3146), 170 µM ascorbate-2-phosphate, 1 mM sodium pyruvate (Biochrom), 350 µM proline (Carl Roth) and 10 ng/mL human recombinant TGF-β3 (Escherichia coli, PeproTech, Hamburg, Germany). The cell suspension was portioned into 96-well plate with round bottoms (polypropylene, Corning #3879 via Sigma-Aldrich) with 200 µL per well corresponding to 1.25 × 105 cells per well. Control pellets were cultured without TGF-β3. Medium was changed every second day. After 28 days, RNA was isolated from the cell pellets as described below. Three pellets were pooled for RNA isolation which was performed in duplicates (six pellets in total). One separate pellet was used for analysis of the glycosaminoglycan-to-DNA ratio.
For donor J, cells isolated with the addition of 3.5 mL heparin did not proliferate substantially after passage 2 (see
Table 2). Therefore, the population doubling time could not be calculated and the cells could not be subjected to osteo- or chondrogenic differentiation (marked by x in Figures 2, 3, 5, 6 and 7).
2.7. RNA Isolation and cDNA Synthesis
Duplicate samples from differentiated cells (osteogenic and adipogenic induction) were lysed in 350 µL buffer RLT (Qiagen, Hilden, Germany), lysates were stored at −80 °C. RNA was subsequently isolated with the RNeasy kit (Qiagen #74106) according to the instructions of the manufacturer including on-column-digestion with DNase I. Synthesis of cDNA was performed from 1 µg of RNA with oligo-dT18-primers according to the manufacturer´s suggestions (Fermentas, Bremen, Germany). Total RNA from chondrogenic pellets was isolated with a homogenizer from Bertin technologies (PreCellys 24 lysis and homogenization; 2 × 30 s, 6000 rpm with 40 s pause) and a Precellys® RNA Kit (732-3122, VWR, Langenfeld, Germany). The isolation was carried out according to the manufacturer’s manual. Synthesis of cDNA was performed with 200 ng of RNA including treatment with DNase I and oligo-dT18-primers according to the manufacturer´s suggestions (Fermentas).
2.8. Histological Staining for Osteogenic and Adipogenic Differentiation
Cells were fixed with 4% (w/v) paraformaldehyde (Carl Roth, Karlsruhe, Germany) in PBS for 30 min at 4 °C. Afterwards cells were washed twice with PBS and stored in PBS at 4 °C until histological staining. Directly before staining cells were washed three times with deionized water (Millipore, Darmstadt, Germany).
Lipid droplets were stained with Oil Red O: The dye was dissolved at 0.5% (w/v) in 60% (v/v) 2-propanol and the solution was filtered. Cells were stained with this solution for 30 min in the dark. The staining solution was removed by three washes with water (Millipore quality).
Calcium ions were stained with Alizarin Red S: Cells were stained with a 1.0% (w/v) aqueous Alizarin Red S (Carl Roth) solution (pH = 4.2, made in-house) for one hour at room temperature. After this, the staining solution was removed by three washes with water (Millipore quality).
2.9. Quantification of Calcium and Phosphate Ions in the Cell Layer
Cells were fixed in 4% (w/v) paraformaldehyde in Tris-buffered saline solution (TBS) for 30 min, washed twice in TBS, and harvested in 1 M HCl using a cell scraper. This mix was incubated at 37 °C for 48 h. Cell debris were removed by centrifugation and the supernatants were collected and stored at −20 °C. The subsequent quantification of calcium and phosphate ions in the cell layer was carried out using the Calcium (CPC) LiquiColor® Test and the QuantiChromTM Phosphate Assay, respectively. Both tests were carried out according to the protocols provided by the respective manufacturer (Stanbio, Boerne, USA and Gentaur Molecular, Aachen, Germany).
2.10. Determination of Glycosaminoglycan-to-DNA Ratio for Chondrogenic Differentiation
The chondrogenic pellets (one pellet per experimental condition) were digested at least 18 h at 60 °C with 0.05 mg/mL papain in 100 mM phosphate buffer containing 10 mM EDTA and 10 mM cysteine, pH 6.5. The DNA was stained with 0.2 µg/mL aqueous solution Hoechst 33258 (Sigma) and the fluorescence was measured immediately with a plate reader (excitation 360 nm; emission 460 nm). After staining of glycosaminoglycans with 0.1 mg/mL 1,9-dimethyl-methylene blue (Sigma-Aldrich; 5 mg 1,9-dimethyl-methylene blue mixed with 152 mg Glycin, 119 mg NaCl, and 4.75 mL 0.1 M HCl and filled up with water to 50 mL) the absorbance was read with a plate reader at 530 nm. DNA and glycosaminoglycan content were measured from the same pellet. The ratio of glycosaminoglycan and DNA was calculated by Excel.
2.11. Transcriptome Analyses
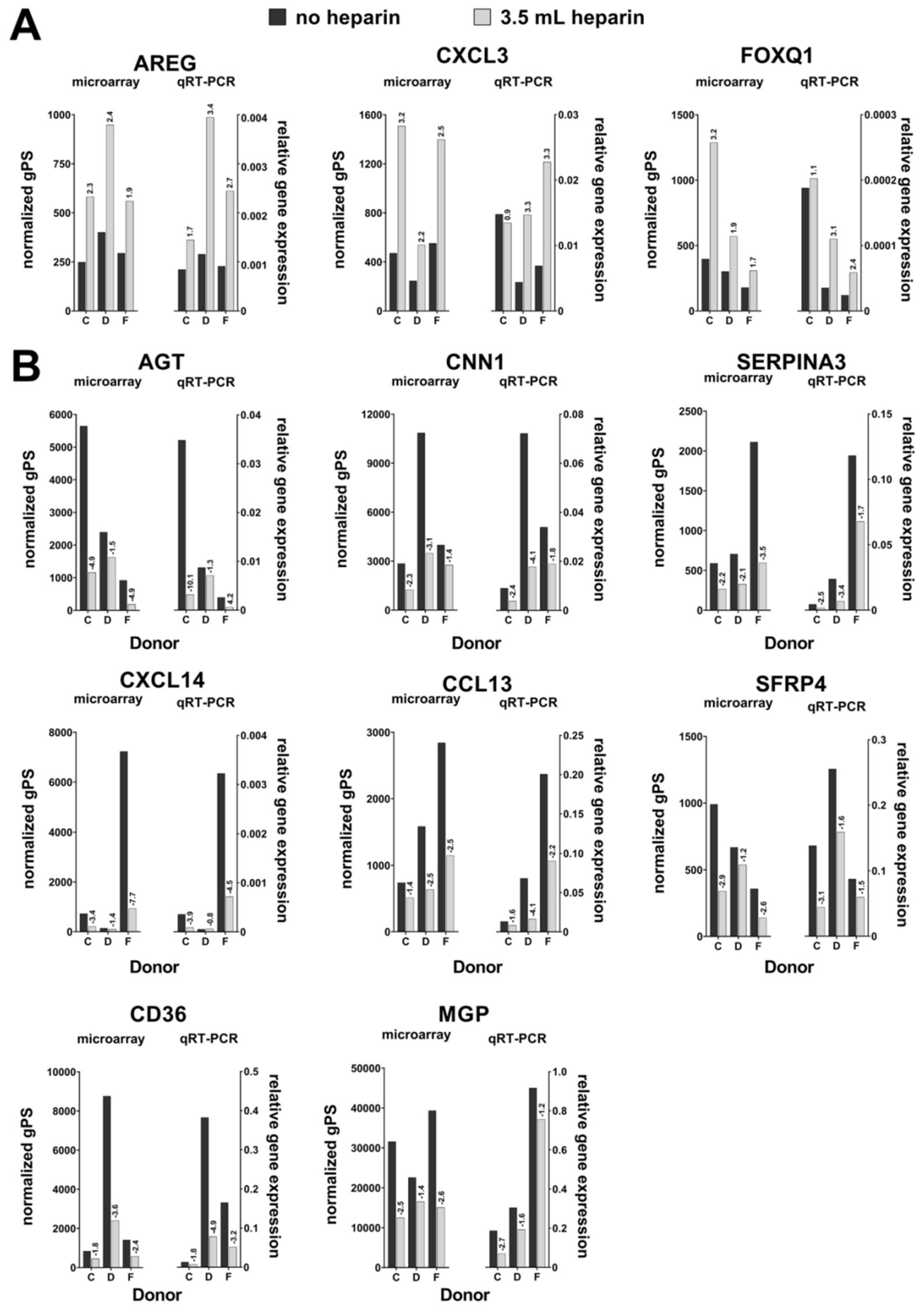
Cells from three different donors (C, D, and F) were randomly chosen. The conditions no heparin and high heparin (called “cell pairs” in the manuscript) were used and RNA obtained at day 0 before induction of osteogenic differentiation was subjected to the analyses.
The Microarray utilized in this study represents a refined version of the Whole Human Genome Oligo Microarray 4 × 44K v2 (Design ID 026652, Agilent Technologies), called “026652QM_RCUG_HomoSapiens” (Design ID 084555) developed by the Research Core Unit Genomics (RCUG) of Hannover Medical School. Microarray design was created at Agilent’s eArray portal using a 1 × 1 M design format for mRNA expression as template. All non-control probes of design ID 026652 have been printed five times within a region comprising a total of 181560 Features (170 columns × 1068 rows). Four of such regions were placed within one 1 M region giving rise to four microarray fields per slide to be hybridized individually (Customer Specified Feature Layout). Control probes required for proper Feature Extraction software operation were determined and placed automatically by eArray using recommended default settings. The array contained 34,127 probes covering a total of 25,992 genes.
100 ng of total RNA were used as input. Synthesis of Cy3-labeled cRNA was performed in ¾ reaction volumes with the “Low Input Quick Amp Labeling Kit One-Color” (#5190-2305, Agilent Technologies) according to the manufacturer’s recommendations. cRNA fragmentation, hybridization and washing steps were carried out as recommended in the “One-Color Microarray-Based Gene Expression Analysis Low Input Quick Amp Labeling Protocol V6.7”, except that 2500 ng of labeled cRNA were used for hybridization.
Slides were scanned on the Agilent Micro Array Scanner G2565CA (pixel resolution 3 µm, bit depth 20).
Data extraction was performed with the “Feature Extraction Software V10.7.3.1” by using the recommended default extraction protocol file “GE1_107_Sep09.xml”.
Measurements of on-chip replicates (quintuplicates) were averaged using the geometric mean of processed fluorescence intensity values of the green channel, “gProcessedSignal” (gPS) to retrieve one resulting value per unique non-control probe. Single Features were excluded from averaging, if they i) were manually flagged, ii) were identified as Outliers by the Feature Extraction Software, iii) lay outside the interval of “1.42× interquartile range” regarding the normalized gPS distribution of the respective on-chip replicate population, or iv) showed a coefficient of variation of pixel intensities per Feature which exceeded 0.5.
Averaged gPS values were normalized by global linear scaling: All gPS values of one sample were multiplied by an array-specific scaling factor. This factor was calculated by dividing a “reference 75th Percentile value” (set as 1500 for the whole series) by the 75th Percentile value of the specific Microarray to be normalized (“Array I” in the formula shown below). Accordingly, normalized gPS values for all samples (microarray data sets) were calculated by the following formula:
Finally, a lower intensity threshold (surrogate value) was defined based on intensity distribution of negative control features. This value was fixed at 15 normalized gPS units. Those measurements which fell below this intensity cutoff were substituted by the respective surrogate value of 15.
Fold changes were calculated as sample with heparin per sample without heparin for the three cell preparations. Genes were selected for verification by quantitative realtime PCR by the following criteria: 1. All probes for the same gene should demonstrate at least twofold changes (up- or downregulation) for at least two cell pairs isolated in the presence of heparin compared to the preparations in the absence of heparin. 2. In case of a smaller fold change, the change for the third cell pair had to be up- or downregulated like for the other two cell pairs. Here, the mean change (expressed as geometric mean of the three ratio values) had to be more than twofold. 3. For downregulated genes, the normalized gPS values had to be >100 for the samples without heparin (i.e., were lower in the samples with heparin). 4. For upregulated genes, the normalized gPS values had to be >100 for the samples with heparin (i.e., were lower in the samples without heparin). The conditions 3 and 4 were chosen to get rid of less reliable measurements nearby the sensitivity limit of the microarray assay. The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus (Edgar et al., 2002) and are accessible through GEO Series accession number GSE145433 (
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE145433).
2.12. Quantitative Realtime RT-PCR
qRT-PCR analyses were performed by using the Applied Biosystems® StepOnePlus instrument (Life Technologies, via Thermo Fisher Scientific). Each sample was measured in duplicates and the mean values were calculated. The gene specific assays as well as the Fast Advanced Mastermix were purchased from Life Technologies: RPS29 (Hs03004310_g1; house-keeping gene), Tissue Non-Specific Alkaline Phosphatase (ALPL, Hs00758162_m1), Osteocalcin (BGLAP, Hs01587814_g1), Bone Sialoprotein (IBSP, Hs00173720_m1), Runt-Related Transcription Factor (RUNX2, Hs00231692_m1), Osteopontin (SPP1, Hs00959010_m1) (all for osteogenic differentiation), Aggrecan (ACAN, Hs00153936_m1), Collagen Type II Alpha I (COL2A1, Hs01064869_m1), Sex Determining Region Y-Box 9 (SOX9, Hs00165814_m1), Matrix Metalloprotease 13 (MMP13, Hs00233992_m1) (all for chondrogenic differentiation), peroxisome proliferator- activated receptor gamma (PPARG, Hs01115513_m1), Fatty Acid Binding Protein 4 (also called adipocyte-FABP or adipocyte protein 2 (aP2), gene name FABP4, Hs00609791_m1) (both for adipogenic differentiation). Amphiregulin (AREG, Hs00950669_m1), Angiotensinogen (AGT, Hs01586213_m1), Calponin, basic (CNN1, Hs00959434_m1), C-C motif chemokine ligand 13 (CCL13, Hs00234646_m1), CD36 (Hs00354519_m1), C-X-C motif chemokine ligand 3 (CXCL3, Hs00171061_m1), C-X-C motif chemokine ligand 14 (CXCL14, Hs01557413_m1), Forkhead Box Q1 (FOXQ1, Hs00536425_s1), Matrix Gla Protein (MGP, Hs00179899_m1), serpin peptidase inhibitor clade A3 (SERPIN A3, Hs00153674_m1), and Secreted Frizzled-Related Protein 4 (SFRP4, Hs00180066_m1) were analyzed as potentially heparin-regulated genes. Analyses were implemented according to the manufacturer´s instructions. Data were evaluated by the delta CT-method with ∆CT = CT (cycle threshold, gene of interest) minus CT (cycle threshold, housekeeper, here RPS29) resulting in relative gene expressions of 2-∆CT.
4. Discussion
In the field of mesenchymal stromal cells, diverse cell populations from many different tissues may be summarized due to their joint minimal features. In the present study, the cell populations were defined as bone marrow-MSCs based on their plastic-adherent growth, the presence or absence of cell surface antigens determined by flow cytometry and their in vitro differentiation. Of special notice, all cell preparations were striking with respect to their CD90 expression, which was notably less than the 95%, which is suggested by the minimal criteria for positive antigens on the MSC surface. We have observed this low level of CD90 surface expression with antibodies against different CD90 epitopes and have also observed that the surface expression of CD90 declines with passaging [
28]. This low CD90 surface expression seems to be favored by the medium used for MSC expansion since it was observed to much less extent in media containing human serum or human platelet lysate and in the absence of FGF-2 (unpublished data).
It becomes more and more relevant that more precise definitions will be necessary to further develop the field in the future. A prominent example from the present study is the cytochemical staining of osteo- or adipogenic differentiation as well as the glycosaminoglycan/DNA ratios of chondrogenic differentiation, which did not correlate with the results of gene expression analyses of the representative marker genes. Only the elevated ratio of calcium to phosphate in the cell layers—indicative of mineralization in form of hydroxyapatite [
29]—correlated with positive Alizarin Red S-staining since both methods quantify calcium ions. However, it is important that cell layers do not detach during investigation of the cell culture. In many cases—in the present study as well as in studies from other groups—floating material consisting of calcium phosphate layers was removed by medium changes during cell culture and prevents accurate determinations of the calcium content. Finally, it is not clear whether faint Alizarin Red S-staining should be taken as evidence for successful differentiation—as with the frequency of washing after removing the stain the intensity declines.
Our data demonstrate that the addition of heparin for anticoagulation of bone marrow during sampling has no influence on the quality of isolated MSCs judged by the minimal criteria and transcriptome analyses. Cells isolated in the absence of heparin and in the presence of high heparin concentrations likewise adhered to tissue culture material as well as no effect during proliferation was observed (assessed by population doubling times). Expression of the cell surface antigens according to the International Society for Cell Therapy minimal criteria were not affected by heparin and neither was the outcome of in vitro trilineage differentiation. Transcriptome analyses were performed in order to obtain a more detailed molecular understanding of the effect of heparin on the isolated cells. Consistent changes of twofold or more were observed in eleven genes only while 25,992 genes were present on the arrays, and the changes in expression levels must be considered as relatively low. These genes were verified by qRT-PCR analyses for nine cell populations with all experimental conditions. Overall, across all tested donors no single gene exhibited a clear tendency of heparin-dependent regulation (
Supplemental Figure S4). These findings deviate from those of earlier publications. However, it is important to remember that in the present study, the effect of heparin addition during bone marrow sampling on subsequent cell performance was assessed; no heparin was added during cell expansion. In the previous studies, isolated cells were expanded and cultured in the presence of heparin, which is a different approach. The results of the previous studies were briefly summarized in the introduction [
11,
12,
14].
Several more genes were detected in the transcriptome analysis, which were regulated in only two of the three investigated donor cell pairs. Therefore, these genes were not verified by qRT-PCR analyses. Anyhow they might be potentially interesting due to the fact that some of these genes function in blood coagulation, which seems reasonable in the context of heparin as a stimulus: Thrombomodulin (upregulated), haptoglobin, and transferrin (downregulated with increasing heparin concentration).
The hemocompatibility of culture-expanded MSCs is an important issue, in particular in in vivo applications using systemic administration. The instant blood-mediated inflammatory reaction (IBMIR) is responsible for compromising the survival and function of systemically applied cells [
30,
31]. In contrast, endothelial cells are largely protected from this reaction since they have strong antithrombotic properties mediated by expression of nitric oxide, prostacyclin, tissue factor pathway inhibitor (gene name:
TFPI), and heparin-bound antithrombin [
31]. One study comparing gene expression in human MSCs and endothelial cells could demonstrate that tissue factor pathway inhibitor (gene name:
TFPI), tissue-type plasminogen activator (gene name:
PLAT), urokinase-type plasminogen activator (gene name:
PLAU), prostacyclin synthase (gene name:
PTIGS, with prostacyclin 2, PGI2, being a strong inhibitor of platelet activation), thrombomodulin (gene name:
THBD), plasminogen activator inhibitor type 1 (gene name:
SERPINE1), and procoagulant platelet factor 4 (gene name:
CXCL4) were similarly expressed in both cell types [
31]. However, strong expression of collagen type I and fibronectin 1 plus weak expression of tissue factor (also called CD142, gene name
F3) on MSCs caused platelet activation and IBMIR [
31]. Consequently, selection of tissue factor-deficient MSCs from bone marrow has been suggested to improve hemocompatibility [
32].
Some of the bone marrow donors in the present study had pre-existing conditions. However, except for one patient (donor K), who had a von Willebrand-syndrome, none of these comorbities is highly likely to affect the coagulation (
Table 1). As pointed out in the introduction, different anti-coagulants are available, which could differentially impact on the behavior of MSCs. Consequently, an analysis comparing different anticoagulants might be performed in the future. In addition, many studies have demonstrated the decisive influence of cell expansion and culture protocols on MSC features. To mention a few: The influence of FCS or FGF-2 (which interacts with heparin). The issue of medium additives is covered in many articles (see, e.g., [
15]). As a matter of fact, apart from composition, concentrations are important. We used a low concentration of 2 ng/mL FGF-2 (compared to 10 or even 20 ng/mL), which has been demonstrated to enhance the proliferative potential [
33], to suppress cellular senescence [
34] and, in these low concentrations, to retain the multilineage differentiation potential of MSCs [
35,
36].
Unfortunately, apart from adhesion to tissue culture plastics, there is no consensus for unequivocal identification and prospective isolation of MSCs, which would be highly important, e.g., the use of CD146-positive cells [
37], perivascular stromal/stem cells [
38], or the human skeletal stem cell characterized by surface expression of Podoplanin, CD73 (Ecto-5’-Nucleotidase) and CD164 (Endolyn) [
39]. Also, MSCs isolated from different tissue sources may have a different developmental origin [
40] and do not necessarily have equivalent biological properties [
4,
41]. The removal of the cells from their original tissue or stem cell niche is an important issue to be covered in future studies since during cell culture, the cells will lose some or many of their important features, which they exhibited in vivo. This also includes their differentiation potential which may notably differ in vitro and in vivo (see, e.g., [
4,
42], or opinion article [
43]). Therefore, finding a way to expand MSCs in high quality with high potential for the desired application is highly important. The criterion of plastic adherence implies that additional cell types with plastic adherent properties might be co-isolated and co-expanded, e.g., macrophages or fibroblasts. One previous study from the present group of researchers has demonstrated that in passage 2 and thereafter, macrophages are not present [
28]. The situation is more complicated with fibroblasts since fibroblasts and MSCs share a lot of surface markers. Consequently, it is important to find some markers, which are only expressed on the surface of MSCs. One group of researchers showed that CD146 was only present on MSCs and CD166 was upregulated in MSCs but not in fibroblasts. Another marker is CD9, which is downregulated in MSCs but upregulated in fibroblasts. However, the authors concluded that “markers distinguishing mesenchymal stem cells from fibroblasts are downregulated with passaging” [
44].
The combination of flow cytometric analysis and high-throughput single cell RNA-sequencing, without in vitro-expansion of cell populations, has recently led to new insights into fibroblast populations. In fact, the fibroblast that was historically described as a connective tissue-cell that secreted proteins, especially collagen, thereby contributing to formation of the extracellular matrix of connective tissue and expressing vimentin [
45], is now revealed as a population of cells with different features. Impressive studies have been conducted on fibroblasts from synovial tissue. In the literature, these cells are often termed “type B synovial lining cells”, fibroblast-like synoviocytes, mesenchymal stromal cells, or other with expression of cell surface markers like CD44 and InterCellular Adhesion Molecule-1, which characterizes these cells into the fibroblast lineage. Specifically, in the lining layer of the synovial tissue, fibroblasts were identified by expression of Fibroblast Activation Protein Alpha [
46] or expression of CD55 [
47]. In the sublining layer, fibroblasts were identified by expression of CD90/THY1 and additional factors [
46,
47] including Podoplanin [
39]. Notably, CD90 is one important marker for MSCs according to the minimal criteria [
1]. Similar to MSCs, studies have shown in vitro-differentiation into mesenchymal cell lineages and also described immunomodulatory properties for fibroblasts (reviewed in [
45]).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}