
Abstract
Introductions of invasive, non-native species in the marine environment are increasing as human activity within coastal areas rises. Genetic datasets are useful tools to identify source populations, track routes of invasions, and illuminate the role of genetic variation in the establishment and subsequent spread of novel introductions. Here, a microsatellite dataset is used to estimate the genetic diversity and population structure of 7 introduced Didemnum vexillum populations in Britain and Ireland, 4 of which are associated with aquaculture and 3 with marinas. Genetic differentiation observed between these populations indicates human-mediated transport as the main mechanism underlying the population structure of D. vexillum in Britain and Ireland. In addition to elucidating patterns of population structure we found that aquaculture sites showed significantly higher genetic diversity (measured as allelic richness) in comparison to the marina sites. We discuss these findings in relation to the history of each invasion, the complex life history of D. vexillum, and available evidence of the relative invasiveness of these populations. Our results show numerous interesting patterns which highlight further research avenues to elucidate the complex factors underlying the global spread of this successful invader.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Examining patterns of genetic diversity in invasive non-native species (INNS) can provide important mechanistic insights into the pathways of invasion and assist future management (Wellband et al. 2017). Historically, newly introduced populations of INNS were expected to exhibit low levels of genetic variation due to genetic drift, a result of population bottlenecks and founder events following transport from their native range (Dlugosch and Parker 2008; Crawford and Whitney 2010). Despite these expectations, many successful introductions of INNS have retained high genetic diversity (Lavergne and Molofsky 2007; Crawford and Whitney 2010; Wellband et al. 2017), likely the result of high propagule pressure (Simberloff 2009), multiple introduction events (e.g., Kolbe et al. 2004; Dlugosch and Parker 2008), and/or stratified dispersal (Darling and Folino-Rorem 2009; Tobin and Blackburn 2008; Berthouly-Salazar et al. 2013). Rius et al. (2015), report clear evidence of reduced genetic diversity in only 23% studies of introduced marine populations, with most studies (74%) reporting no change in diversity between introduced and native populations. In addition to variability in genetic characteristics, inter-specific comparisons of INNS suggests that there is a high degree of variability in the rate of range expansions following new invasions, with some populations expanding rapidly (O’Neill and Dextrase 1994), and others experiencing significant lag phases before expansion (Aikio et al. 2010). Further, not all populations of an INNS have equal potential for becoming invasive (Allendorf and Lundquist 2003), and instances of marine INNS with both invasive and non-invasive populations have been reported (e.g., Osman and Whitlatch 2007).
The carpet sea squirt (Didemnum vexillum) is a colonial ascidian native to Japanese waters (Stefaniak et al. 2012) that has invaded temperate regions globally, likely via the movement of ships with fouled hulls, and/or as epifaunal growth on cultured Pacific oysters. D. vexillum is an invasive species of particular concern due to its ability to significantly alter the structure, biodiversity and function of ecosystems and communities (Mckenzie et al. 2017). Despite reductions of genetic diversity in comparison to native populations in both mitochondrial (Smith et al. 2012, 2015; Ordóñez et al. 2015) and nuclear (Casso et al. 2019a) markers, introduced populations from a single clade of D. vexillum have successfully colonised a variety of environments (Stefaniak et al. 2012; Smith et al. 2015). Across introduced populations, colonies vary in size (Coutts 2002; Valentine et al. 2007), morphology (Coutts and Forrest 2007; Lambert 2009), and in their extent of spread from artificial structures where they were originally introduced to natural habitats. Dispersal within regions where D. vexillum has been introduced is likely a combination of both natural and human-mediated dispersal. Short-distance dispersal likely occurs frequently via larvae (D. vexillum has a short-lived larval duration lasting from 2–36 h; Fletcher et al. 2013), while sporadic long-distance movements can occur both naturally through fragmentation and reattachment of colonies (Morris and Carman 2012) and/or rafting of colonies attached to floating materials (Worcester 1994) and via human-mediated mechanisms.
Increasing economic activity and vessel movement within and around the Irish Sea make the continued spread of D. vexillum a significant threat to both British and Irish ecosystems and economies. Given the complex interaction between natural and human-mediated dispersal, and its range of observed phenotypes, there is still much to learn about D. vexillum introductions, and genetic tools remain invaluable resources to address these questions. Here we estimate genetic differentiation of D. vexillum populations around the coasts of Britain and Ireland. We hypothesize that human-mediated dispersal is most likely to drive the genetic structure between populations and predict that sites connected by human activity (e.g., marina sites connected by vessel movement or aquaculture sites linked by transfer of contaminated seed stock) will be more closely related, regardless of geographic distance. We also estimate genetic diversity for each population, and discuss these results in light of qualitative evidence of invasiveness, and the complex life-history of D. vexillum. If the variable invasiveness of D. vexillum introductions can be linked to specific genetic characteristics (i.e. discrete genetic clusters or patterns in diversity), the management of both existing and novel introductions can be prioritized to those with greater risks of impact.
Methods
Study area
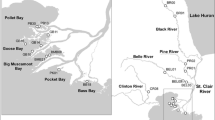
The coastlines of Britain and Ireland have experienced multiple introductions of Didemnum vexillum since it was first recorded in Ireland in 2005 (Malahide Marina; Minchin and Sides 2006) and in Britain in 2008 (Holyhead marina, Wales; Griffith et al. 2009). The current distribution of D. vexillum includes numerous introductions concentrated in areas supporting human activity (oyster aquaculture farms and marinas). Here, we sampled from 7 geographically isolated introduced populations within England, Ireland and Wales (Fig. 1, Table 1). We also qualitatively described each population (colonization history, morphology and habitats colonized) by combining evidence from peer-reviewed and grey literature sources alongside anecdotal evidence from industry and government professionals and our own personal observations (Online Resource 1).

Sampling site locations for Didemnum vexillum introduced populations within the UK and Ireland. Aquaculture and marina sites are represented as white and black circles, respectively. The size of circle is proportional to sample size
Sample collection and DNA extraction
We sampled a total of 419 D. vexillum colonies from seven locations in the summer months between July 2017 and September 2019. Clew Bay, Galway Bay, Dunmanus Bay and Kent are sheltered sites in which oyster aquaculture takes place on intertidal trestles. The sites at Malahide and Holyhead are marinas containing boats and other floating structures (buoys, pontoons, etc.). Strangford Lough supports both oyster aquaculture and marina infrastructure both of which have been colonised by D. vexillum. It is unknown whether the D. vexillum population in the marina that was sampled for this study spread from aquaculture sites or originated from a separate introduction directly to the marina. See Online Resource 2 for detailed information on sampling methods.
Total genomic DNA was extracted from tissue sections using a Qiagen DNeasy Blood & Tissue Kit, according to the manufacturer’s protocol. As D. vexillum colonies have been reported to fuse and form chimeric colonies at the fusion interface (Fidler et al. 2018; Casso et al. 2019b), DNA was extracted from a small section of tissue (< 100 mg) to minimize the probability of capturing a fusion interface.
Genetic profiling of samples
Before microsatellite profiling, we sequenced an ~ 600 bp region of the COI gene in a subset of 71 randomly chosen samples across all populations (between 3 and 22 samples/population) to confirm species ID using the tunicate-specific primers of Stefaniak et al. (2009). As this is the first known report of D. vexillum in Dunmanus Bay, we sequenced all samples collected from this site at the COI gene, in addition to an ~ 480 bp fragment of the 18S rRNA gene using the primers designed by Price et al. (2005).
Twenty microsatellite loci from published literature were initially assessed for use in this study (Abbott et al. 2011; Fidler et al. 2018; Watts et al. 2019), from which we selected 7 polymorphic loci (Dvex03, Dvex10, Dvex11, Dvex18, Dvex19, Dvex20 and Dvex42) that could be amplified reliably and scored consistently across all samples. See Online Resource 2 for detailed information on genetic profiling methods.
Genetic diversity and population structure
The R (R Core Team 2018) package adegenet version 2.0.1 (Jombart 2008) was used to estimate observed (HO) and expected (HE) heterozygosity for each locus, and to conduct t-tests to evaluate whether average HO and HE were significantly different across loci for each population. This package was also used to conduct Bonferroni-corrected (α = 0.05) chi-squared tests to detect loci that departed from Hardy–Weinberg Equilibrium (HWE). GENEPOP (version 4.7; Raymond and Rousset 1995; Rousset 2008) was used to implement Fisher’s exact tests for linkage disequilibrium (LD) for all locus pairs within each population. HP-RARE version 1.1 (Kalinowski 2005) was used to estimate average allelic richness (AR) and private allelic richness for each population, which were corrected for our lowest sample size (N = 15) using rarefaction. Lastly, the R package diversity version 1.9.90 (Keenan et al. 2013) was used to generate multiple estimates of both global and pairwise genetic differentiation (FST [Weir and Cockerham 1984]; GST [Nei 1973]; and Jost’s D [Jost 2008]) in addition to the inbreeding coefficient, FIS, with 95% confidence intervals on each estimate (999 bootstraps).
To determine whether there were significant differences in genetic diversity between populations from different habitats, we conducted comparisons of genetic diversity metrics (HO, AR, gene diversity [HS] and FIS) among populations grouped by habitat type (aquaculture vs. marina) using the software FSTAT v2.9.4 (Goudet 2003). For this analysis we employed two-tailed tests using 10,000 permutations to test for significance. To examine the distribution of genetic variation within and among samples in relation to both population and habitat type, we conducted an analysis of molecular variance (AMOVA) in Arlequin v3.5.2.2 (Excoffier and Lischer 2010) and evaluated the significance of differentiation between and within groups using 10,000 permutations. As the Kent population may have been originally introduced via aquaculture but since spread to other natural and artificial environments, we conducted 2 tests for both of the analyses described above: (1) including Kent in the aquaculture group, and (2) excluding Kent from the analysis.
In addition to estimates of genetic differentiation (FST, GST, Jost’s D), the Bayesian clustering software STRUCTURE version 2.3.4 (Pritchard et al. 2000) was used to test scenarios of K = 1–7, running 10 iterations for each value of K and using a burn-in period of 500,000 followed by 1 × 106 Markov chain Monte Carlo iterations. An admixture model was specified, as secondary introductions or movement of vessels/aquaculture equipment between sites may have facilitated admixture between populations. As STRUCTURE has been shown to incorrectly estimate K when sampling is unbalanced (Wang 2017), the independent allele frequency model was selected, the alternative ancestry prior was applied (allowing for unequal representation of source populations in each sampled population), and the default value of ALPHA was changed to 1/K (0.14). The Evanno method (Evanno et al. 2005) was implemented in STRUCTURE HARVESTER (Earl and vonHoldt 2012) to select the most likely number of genetic clusters, and the 10 replicate runs for the best supported K were summarized with the software CLUMPP (Jakobsson and Rosenberg 2007). The results were visualised with DISTRUCT (Rosenberg 2004). Due to the reliance of STRUCTURE on the underlying assumptions of population genetics models, population structure was also assessed with a multivariate analysis, Discriminant Analysis of Principal Components (DAPC), implemented in the R package adegenet version 2.0.1 (Jombart 2008). As DAPC does not rely on the assumption of mutation-drift-equilibrium, it has been suggested to be appropriate for invasive species (Wellband et al. 2017). It has also been shown to be a reliable method for detecting genetic clines or clusters that were not detected with STRUCTURE in weakly differentiated populations (Jombart et al. 2010; Kanno et al. 2011).
Results
Genetic diversity and population structure
Of our sample subset sequenced at the COI gene (N = 71), all were identified as D. vexillum with 99–100% identity matches to at least 1 D. vexillum sequence in the NCBI GenBank database. At the 18S rRNA gene, all Dunmanus Bay samples were identified as D. vexillum with 99–100% identity matches to at least 1 D. vexillum sequence in the GenBank database. We trimmed sequences to remove base calls of low quality and submitted all sequences to GenBank (Accession Numbers COI: MW425612-MW425681; 18S: MW415990-MW416011).
We observed a total of 6.9% clones in our dataset (range 0–18.5%; Table 1), all of which were collected from the same local area within each site. We also observed very few putative chimeras in our samples (2.7% of all samples) across sites (Table 1). The number of alleles per locus ranged between 3 and 14 within our dataset. See Online Resource 3 for a summary of HO/HE, HWE, LD, and FIS results. Allelic richness (AR) was lower in marina sites (average AR = 3.15) compared to aquaculture sites (average AR = 4.64; Table 2). Sites with the highest AR were also those that showed the highest numbers of private alleles (Table 2). Comparisons of genetic diversity metrics (HO, AR, gene diversity and FIS) for populations grouped by habitat revealed a significant difference in AR between aquaculture and marina habitats, regardless of whether Kent was included (p = 0.025) or excluded (p = 0.025) from the analysis. All other tests were insignificant (p > 0.05).
Estimates of genetic differentiation showed largely similar patterns for each of the metrics measured (FST, GST, and Jost’s D) and only FST and Jost’s D are reported here (Online Resource 4). For both FST and Jost’s D, genetic differentiation was significant for global estimates (global FST = 0.084 [95% CI’s 0.073–0.093]; global Jost’s D = 0.109 [95% CI’s 0.091–0.133]), and all pairwise population comparisons with the exception of Clew Bay and Dunmanus Bay. The most highly differentiated sites were marina sites, with Strangford Lough being the most differentiated across all sites. While Holyhead Marina was closely related to Malahide Marina, it was also highly differentiated from all other sites. Of the aquaculture sites, Galway Bay was most highly differentiated, with the lowest differentiation between Clew Bay and Dunmanus Bay. Our AMOVA analysis revealed significant partitions in genetic variation among habitat groups, among populations within habitat groups, among individuals within populations and within individuals, the latter explaining the largest percentage of variation in our dataset (Online Resource 5). These significant differences were retained regardless of whether we included Kent in the analysis, however we note that the difference among habitat groups becomes less significant when Kent is included (p < 0.001 with Kent removed vs. p < 0.01 with Kent included).
Both Evanno’s Delta K and Ln(K) plots provided strong support for 3 genetic clusters using our set of 7 presumably neutral microsatellite markers (Online Resource 6). These clusters corresponded to groupings of (1) marina (Holyhead and Malahide Marinas) and (2) aquaculture (Galway Bay) sites, with (3) Kent and Strangford Lough forming a third, genetically distinct cluster (Fig. 2). The aquaculture sites Clew Bay and Dunmanus Bay showed admixture between genetic clusters 2 and 3. The results of the DAPC analysis mirrored the STRUCTURE results closely (Fig. 3).

STRUCTURE plot based on 10 replicates representing the proportion of an individual’s genome assigned to one of three genetic clusters identified across 7 introduced populations of the invasive marine tunicate Didemnum vexillum within the UK and Ireland. Here we show results for K = 2 (top), K = 3 (middle), and K = 4 (bottom), with strong support for K = 3 as the number of genetic clusters explained by the data

Axes 1 and 2 of a Discriminant Analysis of Principal Components (DAPC) plot showing genetic clustering of 378 colonies of the invasive tunicate Didemnum vexillum sampled across 7 introduced populations within the UK and Ireland. Population labels are positioned at approximately the centroid of each cluster, with slight adjustments made to prevent label overlap
Discussion
Estimates of genetic relatedness among D. vexillum populations show a complex picture, with a combination of effects related to regional proximity and habitat type. Firstly, AMOVA results suggest significant partitions in genetic variation among habitat types (i.e., marinas vs. aquaculture), even though these groupings account for the smallest proportion of variation overall. Additionally, while we show genetic connectivity between some spatially clustered sites, there are notable exceptions of long-distance connectivity, providing evidence for potential source populations and routes of invasion within Britain and Ireland. For example, high genetic connectivity was detected between Malahide and Holyhead marinas, situated on opposite sides of the Irish Sea with substantial boat traffic between them. In contrast, the third marina site in Strangford Lough, on the east coast of Northern Ireland, is genetically divergent from the other two marinas but shows high connectivity to Kent, a site associated with aquaculture on the southeastern coast of England. While the pathways leading to D. vexillum introduction at these sites are not well understood, movement of oysters between Kent and Strangford Lough for aquaculture offers a possible explanation. Given the limited natural larval dispersal capabilities of D. vexillum (Fletcher et al. 2013), the patterns of genetic differentiation and population structure reported here (Figs. 2, 3, Online Resource 4) are likely to reflect human-mediated dispersal rather than natural dispersal as the primary mode of connectivity between populations throughout Britain and Ireland. This finding agrees with a growing body of literature attributing aquaculture practices and maritime trade to the introduction and spread of INNS globally (e.g., Voisin et al. 2005; de Barros et al. 2009; Blakeslee et al. 2010; Meistertzheim et al. 2013; Cruz Capel et al. 2017).
At the local level, natural and human-mediated dispersal are more difficult to disentangle. The presence of individuals from STRUCTURE cluster 2 in all three aquaculture-associated populations on the west coast of Ireland (Galway Bay, Clew Bay and Dunmanus Bay) suggests a common source. However, it is not clear whether this has arisen due to natural dispersal from a single introduction or separate human-mediated introductions (i.e., the use of oyster seed from a common contaminated source at all three sites). The capabilities of D. vexillum colonies to fragment and re-attach to new substrates supports the potential for natural dispersal and connectivity between proximate populations (Bullard et al. 2007; Reinhardt et al. 2012) and as fragments can be viable in the water column for up to 3 weeks (Morris and Carman 2012), dispersal of D. vexillum via drifting or rafting may promote connectivity at a regional scale (i.e., among aquaculture sites). In contrast, individuals assigned to STRUCTURE cluster 3 are common at Clew Bay and Dunmanus Bay but very rare at Galway Bay, despite the fact that Galway Bay lies between the other two populations. This suggests separate introductions facilitated by human-mediated transportation, or divergence between sites following a single introduction due to a lack of gene flow, and refutes the likelihood of natural dispersal between sites. The inclusion of additional invasive (European) and native (Japanese) populations outside our study area would help to resolve the complex relationships between these populations (e.g. as in Casso et al. 2019a), and importantly allow for the identification of source populations.
In addition to enhancing our understanding of the patterns of connectivity among populations, our work also provides insight into the potential role of environment and/or genetic diversity in determining invasiveness. Qualitative measures of invasiveness were higher in populations of D. vexillum at aquaculture sites, and these populations also had greater allelic richness. Local environment can have an important role in determining the likelihood of INNS becoming invasive (e.g., Alpert et al. 2000; Burns 2006). For example, in the relatively open coastal habitats used for oyster aquaculture, D. vexillum may experience greater competitive advantage (Osman and Whitlatch 2007) and there is greater opportunity for natural dispersal of larvae and therefore local spread. However, marina environments, despite being more confined, may provide superior conditions for growth (e.g., protection from air exposure/desiccation, sunlight, sedimentation, and strong wave action; Daniel and Therriault 2007 and references therein), and opportunities for human-mediated dispersal.
An alternative explanation for the observed variation in invasiveness is the potential for a relationship between genetic diversity and invasive success. Lower genetic diversity can lead to the reduced success of invasive populations via both neutral and adaptive processes that vary temporally. From a neutral perspective, low genetic diversity may be indicative of introductions with low propagule pressure, which can negatively affect invasion success via both genetic and demographic processes (Hufbauer et al. 2013; Blackburn et al. 2015; Bock et al. 2015). For example, populations with lower genetic diversity are more likely to suffer the negative effects of inbreeding depression, which is likely to affect populations at early stages of introduction when population sizes are small (Charlesworth and Charlesworth 1987). Additionally, populations with high genetic diversity have a higher probability of containing genotypes that facilitate establishment success (e.g. Gamfeldt et al. 2005), or resilience to disturbance or novel environments (e.g. Reusch et al. 2005; Crawford and Whitney 2010). Over longer time-scales, admixture may play a large role in determining invasive success (e.g., Keller and Taylor 2010; Hudson et al. 2020), by increasing genetic variation for evolution, creating novel phenotypes via new allele and gene combinations, facilitating heterosis, and masking or purging deleterious mutations which may counteract the initial negative effects of bottlenecks and/or inbreeding (Verhoeven et al. 2011; Bock et al. 2015). Thus, if aquaculture populations have experienced more inter-population admixture than marina populations, this could explain their elevated levels of genetic diversity and increased invasive success in comparison to our more geographically isolated marina populations. From a selection standpoint, if neutral and functional nuclear diversity are positively correlated, then high standing genetic diversity may benefit selection and facilitate survival and spread in novel and stressful environments (Prentis et al. 2008). Indeed, greater genetic variation in ecologically important traits has been related to greater niche breadth in geographically widespread species (Sheth and Angert 2014). While potentially comprising an important mechanism of invasiveness, the influence of selection in D. vexillum cannot be properly investigated with our dataset of neutral markers, and we encourage further research using quantitative traits and functional genomic loci.
As a group, the three marina populations analysed here exhibited significantly lower allelic richness than the four populations associated with oyster aquaculture. While higher genetic diversity in aquaculture populations may be attributed to the greater scale of sampling and more diverse substrates sampled at aquaculture sites in comparison to marinas, we note that our sampling was representative of true differences between the habitat types; marina populations in general did not show extensive spread throughout the local area and did not colonise available natural substrates. Given that the introduction of D. vexillum within the UK and Ireland is spatially distributed such that populations associated with similar habitats (i.e. aquaculture vs. marina) are spatially clustered, the extent to which we are able to disentangle the relative contribution of habitat type from other regional biotic and abiotic factors is limited.
While genetic datasets can be useful to describe underlying patterns of connectivity and diversity in INNS populations, the complex ecology of many INNS make interpretations from genetic data difficult. D. vexillum undergoes both sexual and asexual reproduction (Fletcher et al. 2013), which can make interpretations of links between neutral genetic diversity and invasiveness difficult (Dlugosh et al. 2015). Uniparentally reproducing species may promote invasiveness if (1) certain clonal lineages are better adapted, (2) there is a shift in reproductive mode (Le Cam et al. 2020; Wellband et al. 2017) or, (3) quantitative genetic variation associated with polygenic traits is retained despite bottlenecks or founder events (Barrett 2015). Our data shows the highest proportion of clonal reproduction in our seemingly least invasive marina population, thereby contrasting with the hypothesis that clonal reproduction should increase invasive success. However, the high proportion of unique colonies included in our dataset suggest that there is a strong influence of multiple introductions and/or sexual reproduction in our populations. The tendency of D. vexillum colonies to fuse, forming chimeras, further complicates the possible interpretations of our dataset. For example, Watts et al. (2019) show that changes in allele frequencies produced disparate genetic groupings when comparing datasets that included versus those that excluded genetic data from chimeric colonies. Evidence of chimeras was low in this dataset (2.7%) in contrast to other regions (Clancy 2015; Watts et al. 2019; Casso et al. 2019b) suggesting that the impact of chimeras on estimations of genetic clusters would be small in our study area.
While some of the genetic mechanisms involved in the success of INNS in general (Stapley et al. 2015; Tepolt and Palumbi 2015; Hawes et al. 2018a; Gleason 2019), and D. vexillum, specifically [e.g., plasticity (Ordóñez et al. 2015), epigenetics (Hawes et al. 2018b, 2019), and microbiome effects (Casso et al. 2020)], have been identified, many questions remain unanswered. Our results highlight the importance of genetic tools in invasion management, in particular for tracking secondary spread and evaluating the risk of invasiveness in new introductions.
Availability of data and material
The microsatellite dataset generated during the current study is available in the PURE repository, https://doi.org/10.20391/049bdaa6-8ecd-4275-8f2a-f91e834837b3. COI and 18S rRNA sequences are available on GenBank, Accession Numbers MW425612-MW425681 and MW415990-MW416011, respectively.
References
Abbott CL, Ebert D, Tabata A, Therriault TW (2011) Twelve microsatellite markers in the invasive tunicate, Didemnum vexillum, isolated from low genome coverage 454 pyrosequencing reads. Conserv Genet Resour 3:79–81
Aikio S, Duncan RP, Hulme PE (2010) Lag-phase in alien plant invasions: separationg the facts from the artefacts. Oikos 119:370–378
Allendorf FW, Lundquist LL (2003) Introduction: population biology, evolution and control of invasive species. Conserv Biol 17:24–30
Alpert P, Bone E, Holzapfel C (2000) Invasiveness, invasibility and the role of environmental stress in the spread of non-native plants. Perspect Plant Ecol Evol Syst 3:52–66
Barrett SCH (2015) Foundations of invasion genetics: the Baker and Stebbins legacy. Mol Ecol 24:1927–1941
Berthouly-Salazar C, Hui C, Blackburn TM, Gaboriaud C, van Rensburg BJ, van Vuuren BJ, Le Roux JJ (2013) Long-distance dispersal maximizes evolutionary potential during rapid geographic range expansion. Mol Ecol 22:5793–5804
Blackburn TM, Lockwood JL, Cassey P (2015) The infleunce of numbers on invasion success. Mol Ecol 24:1942–1953
Blakeslee AMH, McKenzie CH, Darling JA, Byers JE, Pringle JM, Roman J (2010) A hitchikers guide to the Maritimes: anthropogenic transport facilitates long-distance dispersal of an invasive marine crab to Newfoundland. Divers Distrib 16:879–891
Bock DG, Caseys C, Cousens RD, Hahn MA, Heredia SM, Hubner S, Turner KG (2015) What we still don’t know about invasion genetics. Mol Ecol 24:2277–2297
Bullard SG, Sedlack B, Reinhardt JF, Litty C, Gareau K, Whitlatch RB (2007) Fragmentation of colonial ascidians differences in reattachment capability among species. J Exp Mar Biol Ecol 342:166–168
Burns JH (2006) Relatedness and environment affect traits associated with invasive and noninvasive introduced commelinaceae. Ecol Appl 16:1367–1376
Casso M, Turon X, Pascaul M (2019a) Single zooids, multiple loci: independent colonisations revealed by population genomics of a global invader. Biol Invasions 21:3575–3592
Casso M, Tagliapietra D, Turon X, Pascual M (2019b) High fusability and chimera prevalence in an invasive colonial ascidian. Sci Rep 9:15673
Casso M, Turon M, Marco N, Pascual M, Xavier T (2020) The microbiome of the worldwide invasive ascidian Didemnum vexillum. Front Mar Sco 7:201
Charlesworth D, Charlesworth B (1987) Inbreeding depression and its evolutionary consequences. Annu Rev Ecol Syst 18:237–268
Clancy D (2015) Examining genetic diversity and fusion abilities of an invasive colonial ascidian. Dissertation, San Francisco State University
Coutts ADM (2002) A biosecurity investigation of a barge in the Malborough Sounds. Nelson, New Zealand: Cawthorn Institute, 68 (Cawthorn Report No. 744)
Coutts ADM, Forrest BM (2007) Development and application of tools for incursion response: lessons learned from the management of the fouling pest Didemnum vexillum. J Exp Mar Biol Ecol 342:154–162
Crawford K, Whitney K (2010) Population genetic diversity influences colonization success. Mol Ecol 19:1253–1263
Cruz Capel KC, Toonen RJ, Rachis CTCC, Creed JC, Kitahara MV, Forsman Z, Zilerberg C (2017) Clone wars: asexual reproduction dominates in the invasive range of Tubastraea spp. (Anthozoa: Scleractinia) in the South-Atlantic Ocean. PeerJ 5:e3873
Daniel KS, Therriault TW (2007) Biological synopsis of the invasive tunicate Didemnum sp. Canadian Manuscript Report of Fisheries and Aquatic Sciences 2788, p 53
Darling JA, Folino-Rorem NC (2009) Genetic analysis across different spatial scales reveals multiple dispersal mechanisms for the invasive hydrozoan Cordylophora in the Great Lakes. Mol Ecol 18:4827–4840
de Barros RC, da Rocha RM, Pie MR (2009) Human-mediated global distribution of Styrls plicata (Tunicata, Ascidiacea). Aqaut Invasions 4:45–57
Dlugosch KM, Parker IM (2008) Founding events in species invasions: genetic variation, adaptive evolution, and the role of multiple introductions. Mol Ecol 17:431–449
Dlugosh KM, Anderson SR, Braasch J, Cang FA, Gillette HD (2015) The devil is in the details: genetic variation in introduced populations and its contributions to invasion. Mol Ecol 24:2095–2111
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Res 10:564–567
Fidler AE, Bacq-Labreuil A, Rachmilovitz E, Rinkevich B (2018) Efficient dispersal and substrate acquisition traits in a marine invasive species via transient chimerism and colony mobility. PeerJ 6:e5006
Fletcher LM, Forrest BM, Bell JJ (2013) Natural dispersal mechanisms and dispersal potential of the invasive ascidian Didemnum vexillum. Biol Invasions 15:627–643
Gamfeldt L, Wallén J, Jonsson PR, Berntsson KM, Havenhand JN (2005) Increasing intraspecific diversity enhances settling success in a marine invertebrate. Ecology 86:3219–3224
Gleason LU (2019) Applications and future directions for population transcriptomics in marine invertebrates. Curr Mol Biol Rep 5:116–127
Goudet J (2003) FSTAT (ver. 2.9.4), a program to estimate and test population genetics parameters. http://www.unil.ch/izea/softwares/fstat.html. Updated from Goudet 1995
Griffith K, Mowat S, Holt RHF, Ramsay K, Bishop JDD, Lambert G, Jenkins SR (2009) First records in Great Britain of the invasive colonial ascidian Didemnum vexillum Knott, 2002. Aquat Invasions 4:581–590
Hawes NA, Tremblay LA, Pochon X, Dunphy B, Fidler AE, Smith KF (2018b) Effects of temperature and salinity stress on DNA methylation in a highly invasive marine invertebrate, the colonial ascidian Didemnum vexillum. PeerJ 6:e5003
Hawes NA, Fidler AE, Tremblay LA, Pochon X, Dunphy BJ, Smith KF (2018a) Undestanding the role of DNA methylation in successful biological invasions: a review. Biol Invasions 20:2285–2300
Hawes NA, Amadoru A, Tremblay LA, Pochon X, Dunphy B, Fidler AE, Smith KF (2019) Epigenetic patterns associated with an ascidian invasion: a comparison of closely related clades in their native and introduced ranges. Sci Rep 9:14275
Hudson J, Johannesson K, McQuaid CD, Rius M (2020) Secondary contacts and genetic admixture shape colonization by an amphiatlantic epibenthic invertebrate. Evol Appl 13:600–612
Hufbauer RA, Rutschmann A, Serrate B, Vermeil de Conchard H, Facon B (2013) Role of propagule pressure in colonization success: distentagling the relative importance of demographic, genetic and habitat effects. J Evol Biol 26:1691–1699
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806
Jombart T (2008) adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405
Jombart T, Devillard S, Balloux F (2010) Discriminant analysis of principal components: a method for the analysis of genetically structured populations. BMC Genet 11:94
Jost L (2008) GST and its relatives do not measure differentiation. Mol Ecol 17:4015–4026
Kalinowski ST (2005) HP-RARE: a computer program fro performing rarefaction on measures of allelic diversity. Mol Ecol Notes 5:187–189
Kanno Y, Vokoun JC, Letcher BH (2011) Fine-scale population structure and riverscape genetics of brook trout (Salvenlinus fontinalis) distributed continuously along headwater channel networks. Mol Ecol 20:3711–3729
Keenan K, McGinnity P, Cross TF, Crozier WW, Prodöhl PA (2013) diveRsity: an R package for the estimation and exploration of population genetics parameters and their associated errors. Methods Ecol Evol 4:782–788
Keller LF, Taylor DR (2010) Genomic admixture increases fitness during a biological invasion. J Evol Biol 23:1720–1731
Kolbe JJ, Glor RE, Schettino LR, Lara AC, Larson A, Losos JB (2004) Genetic variation increases during biological invasion by a Cuban lizard. Nature 431:177–181
Lambert G (2009) Adventures of a sea squirt sleuth: unravelling the identity of Didemnum vexillum, a global ascidian invader. Aquat Invasions 4:5–28
Lavergne S, Molofsky J (2007) Increased genetic variation and evolutionary potential drive the success of an invasive grass. Proc Natl Acad Sci USA 104:3883–3888
Le Cam S, Daguin-Thiébaut C, Bouchemousse S, Engelen AH, Mieszkowska N, Viard F (2020) A genome-wide investigation of the worldwide invader Sargassum muticum shows high success albeit (almost) no genetic diversity. Evol Appl 13:500–514
McKenzie CH, Reid V, Lambert G, Matheson K, Minchin D, Pederson J, Brown L et al (2017) Alien species alert: Didemnum vexillum Knott, 002: invasion, impact and control. ICES Cooperative Report No. 335, p 33
Meistertzheim A-L, Arnaud-Haond S, Boudry P, Thébault M-T (2013) Genetic structure of wild European populations of the invasive Pacific oyster Crassostrea gigas due to aquaculture practices. Mar Biol 160:453–463
Minchin D, Sides E (2006) Appearance of a cryptogenic tunicate, a Didemnum sp. fouling marina pontoons and leisure craft in Ireland. Aquat Invasions 1:143–147
Morris JA, Carman MR (2012) Fragment reattachment, reproductive status, and health indicators of the invasive colonial tunicate Didemnum vexillum with implications for dispersal. Biol Invasions 14:2133–2140
Nei M (1973) Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci USA 70:3321–3323
O’Neill CR, Dextrase A, (1994) The zebra mussel: its origins and spread in North America. Proceedings of the fourth international Zebra Mussel conference. Electric Power Research Institute, Palo Alto, California
Ordóñez V, Pascual M, Fernández-Tejedor M, Pineda MC, Tagliapietra D, Turon X (2015) Ongoing expansion of the worldwide invader Didemnum vexillum (Ascidiacea) in the Mediterranean Sea: high plasticity of its biological cycle promotes establishment in warm waters. Biol Invasions 17:2075–2085
Osman RW, Whitlatch RB (2007) Variation in the ability of Didemnum sp. to invade established communities. J Exp Mar Biol Ecol 342:40–53
Prentis PJ, Wilson JRU, Dormontt EE, Richardson DM, Lowe AJ (2008) Adaptive evolution in invasive species. Trends Plant Sci 13:288–294
Price A, Collie JS, Smith DC (2005) 18s ribosomal RNA and cytochrome oxidase gene sequences of Didemnum sp., an invasive colonia tunicate. GSO Technical Report No. 2006–01, Summer Undergraduate Research Fellowship Program in Oceanography, University of Rhode Island, Graduate School of Oceanography, Narragansett, RI, pp 46–53
Pritchard J, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
R Core Team (2018) R: a language and environment for statistical computing. R Foundation for satatistical computing, Vienna, Austria. https://www.R-project.org/
Raymond M, Rousset F (1995) GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. J Hered 86:248–249
Reinhardt JF, Gallagher KL, Stefaniak LM, Nolan R, Shaw MT, Whitlatch RB (2012) Material properties of Didemnum vexillum and prediction of tendril fragmentation. Mar Biol 159:2875–2884
Reusch TBH, Ehlers A, Hämmerli A, Worm B (2005) Ecosystem recovery after climatic extremes enhanced by genotypic diversity. Proc Natl Acad Sci USA 102:2826–2831
Rius M, Turon X, Bernardi G, Volckaert FAM, Viard F (2015) Marine invasion genetics: from spatio-temporal patterns to evolutionary outcomes. Biol Invasions 17:869–885
Rosenberg NA (2004) DISTRUCT: a program for the graphical display of population structure. Mol Ecol Notes 4:137–138
Rousset F (2008) Genepop ‘007: a complete reimplementation of the Genepop software for Windowd and Linux. Mol Ecol Resour 8:103–106
Sheth SN, Angert AL (2014) The evolution of environmental tolerance and range size: a comparison of geographically restricted and widespread Mimulus. Evol 68:2917–2931
Simberloff D (2009) The role of propagule pressure in biological invasions. Annu Rev Ecol Evol Syst 40:81–102
Smith KF, Stefaniak L, Saito Y, Gemmill CEC, Cary SC, Fidler AE (2012) Increased inter-colony fusion rates are associated with reduced COI haplotype diversity in an invasive colonial ascidian Didemnum vexillum. PLoS ONE 7:e30473
Smith KF, Abbott CL, Saiot Y, Fidler AE (2015) Comparison of whole mitochondrial genome sequences from two clades of the invasive ascidian, Didemnum vexillum. Mar Genomics 19:75–83
Stapley J, Santure AW, Dennis SR (2015) Transposable elements as agents of rapid adaptation may explain the genetic paradox of invasive species. Mol Ecol 24:2241–2252
Stefaniak L, Lambert G, Gittenberger A, Zhang H, Lin S, Whitlatch RB (2009) Genetic conspecificity of the worldwide populations of Didemnum vexillum Knott, 2002. Aquat Invasions 4:29–44
Stefaniak L, Zhang H, Gittenberger A, Smith K, Holsinger K, Lin S, Whitlatch RB (2012) Determining the native region of the putatively invasive ascidian Didemnum vexillum Knott, 2002. J Exp Mar Biol Ecol 422–423:64–71
Tepolt CK, Palumbi SR (2015) Transcriptome sequencing reveals both neutral and adaptive genome dynamics in a marine invader. Mol Ecol 24:4145–4158
Tobin PC, Blackburn LM (2008) Long-distance dispersal of the gypsy moth (Lepidoptera: Lymantriidae) facilitated its initial invasion of Wisconsin. Environ Entomol 37:87–93
Valentine PC, Collie JS, Reid RN, Asch RG, Guida VG, Blackwood DS (2007) The occurrence of the colonial ascidian Didemnum sp. on Georges Bank gravel habitat—ecological observations and potential effects on groundfish and scallop fisheries. J Exp Mar Biol Ecol 342:179–181
Verhoeven KJF, Macel M, Wolfe LM, Biere A (2011) Population admixture, biological invasions and the balance between local adaptation and inbreeding depression. Proc Royal Soc B 278:2–8
Voisin M, Engel CR, Viard F (2005) Differential shuffling of native genetic diversity across introduced regions in a brown alga: aquaculture vs. maritime traffic effects. Proc Natl Acad Sci USA 102:15
Wang J (2017) The computer program STRUCTURE for assigning individulas to populations: easy to use but easier to misuse. Mol Ecol Resour 17:981–990
Watts AM, Hopkins GA, Sharyn J (2019) Chimerism and population dieback alter genetic inference related to invasion pathways and connectivity of biofouling populations on artificial substrata. Ecol Evol 9:3089–3104
Weir BS, Cockerham C (1984) Estimating F-statistics for the analysis of population structure. Evol 38:1358–1370
Wellband KW, Pettitt-Wade H, Fisk AT, Heath DD (2017) Differential invasion success in aquatic invasive species: the role of within- and among-population genetic diversity. Biol Invasions 19:2609–2621
Acknowledgements
The authors would like to acknowledge all aquaculture farm and marina operators and owners for their co-operation in facilitating sample collection. We thank Tim Mackie, Jennifer Coughlan, Paul Brooks, Holly Date and Thea Moule who helped in the sample collection for this work, and Caron Evans who ran our samples for genotyping. We are grateful to Mary Hannan (Bord Iascaigh Mhara, Aquaculture), Peter Donlon (Bord Iascaigh Mhara, Aquaculture) and Tim Mackie (Department of Agriculture, Environment and Rural Affairs, Marine and Fisheries Division) who provided information to assist our qualitative assessments of invasiveness. We also thank all anonymous reviewers whose thorough and thoughtful reviews greatly improved this manuscript. Funding was provided by European Regional Development Fund (Grant no. 80939).
Funding
This research was funded by the Ecostructure project (part-funded by the European Regional Development Fund through the Ireland Wales Cooperation Programme 2014–2020).
Author information
Authors and Affiliations
Contributions
All authors conceived the ideas and designed the methodology. MBP, SRV and JEI collected samples. MBP performed the laboratory work and analysed the genetic data. SRV and MBP performed the qualitative assessment of invasiveness. MBP and SRV drafted the manuscript. All authors contributed critically to the drafts and gave final approval for publication.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Prentice, M.B., Vye, S.R., Jenkins, S.R. et al. Genetic diversity and relatedness in aquaculture and marina populations of the invasive tunicate Didemnum vexillum in the British Isles. Biol Invasions 23, 3613–3624 (2021). https://doi.org/10.1007/s10530-021-02615-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10530-021-02615-3