


Genes 2024, 15(5), 549; https://doi.org/10.3390/genes15050549 (registering DOI) - 25 Apr 2024
Abstract
Recent evidence suggests that human gene promoters display gene expression regulatory mechanisms beyond the typical single gene local transcription modulation. In mammalian genomes, genes with an associated bidirectional promoter are abundant; bidirectional promoter architecture serves as a regulatory hub for a gene pair
[...] Read more.
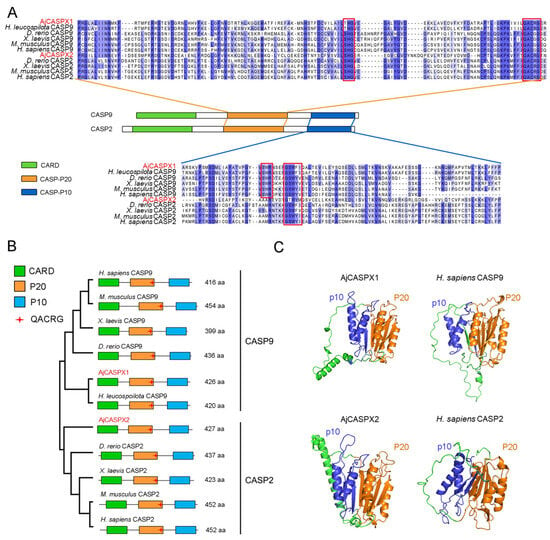
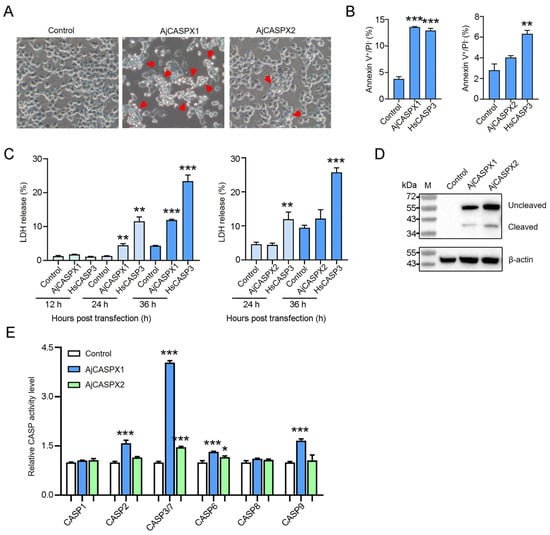
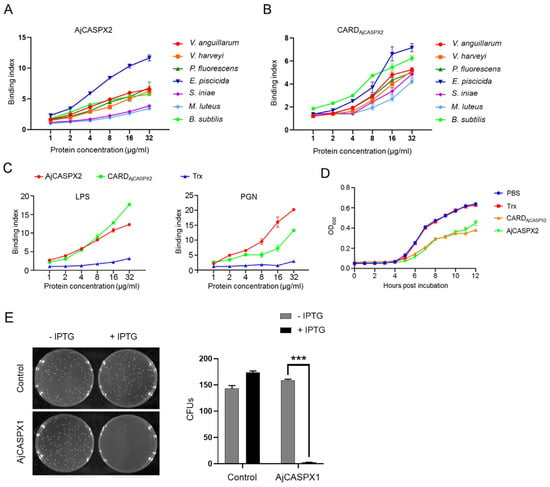
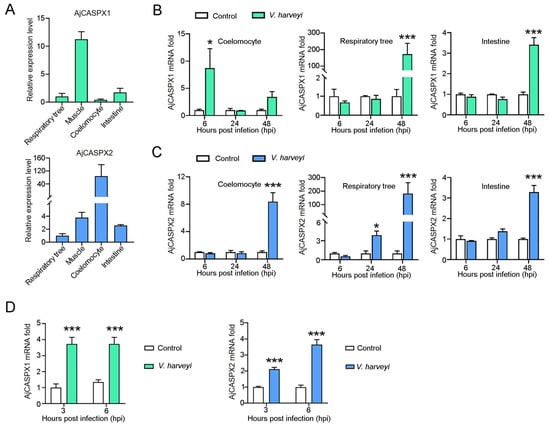
Recent evidence suggests that human gene promoters display gene expression regulatory mechanisms beyond the typical single gene local transcription modulation. In mammalian genomes, genes with an associated bidirectional promoter are abundant; bidirectional promoter architecture serves as a regulatory hub for a gene pair expression. However, it has been suggested that its contribution to transcriptional regulation might exceed local transcription initiation modulation. Despite their abundance, the functional consequences of bidirectional promoter architecture remain largely unexplored. This work studies the long-range gene expression regulatory role of a long non-coding RNA gene promoter using chromosome conformation capture methods. We found that this particular bidirectional promoter contributes to distal gene expression regulation in a target-specific manner by establishing promoter–promoter interactions. In particular, we validated that the promoter–promoter interactions of this regulatory element with the promoter of distal gene BBX contribute to modulating the transcription rate of this gene; removing the bidirectional promoter from its genomic context leads to a rearrangement of BBX promoter–enhancer interactions and to increased gene expression. Moreover, long-range regulatory functionality is not directly dependent on its associated non-coding gene pair expression levels.
Full article
(This article belongs to the Section Molecular Genetics and Genomics)





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}