





Genes 2024, 15(4), 504; https://doi.org/10.3390/genes15040504 - 17 Apr 2024
Abstract
The green monkey Chlorocebus sabaeus, L. 1766, native to West Africa, was introduced to the Cabo Verde Archipelago in the 16th century. Historical sources suggest that, due to the importance of Cabo Verde as a commercial entrepôt in the Atlantic slave trade,
[...] Read more.
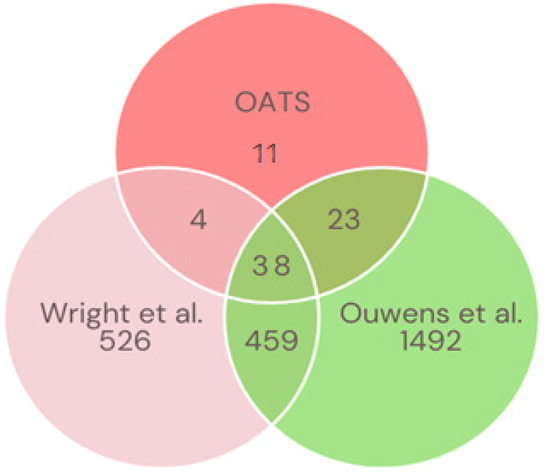
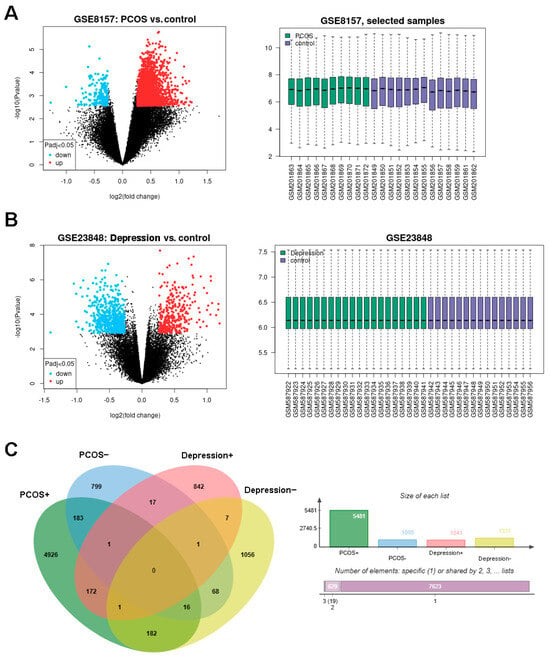
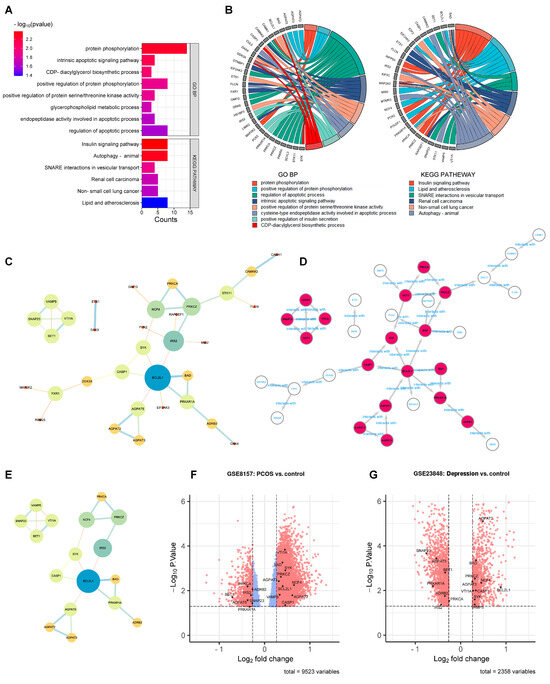
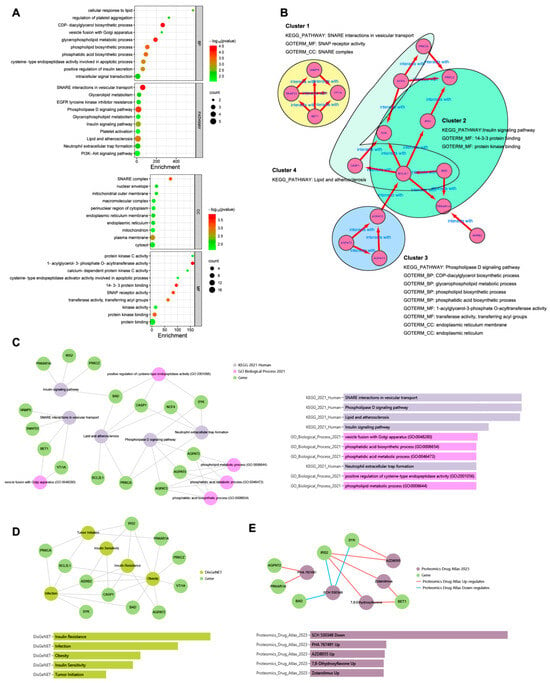
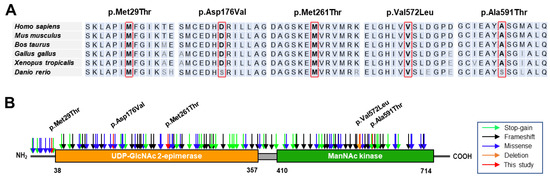
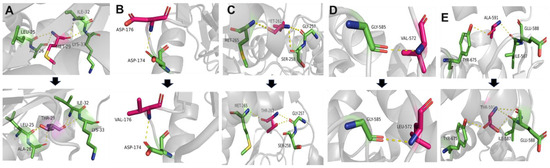
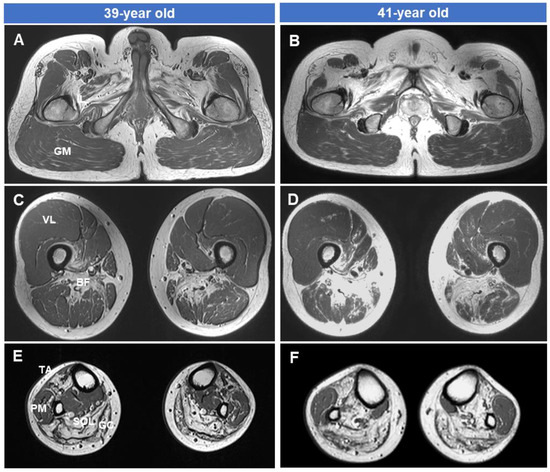
The green monkey Chlorocebus sabaeus, L. 1766, native to West Africa, was introduced to the Cabo Verde Archipelago in the 16th century. Historical sources suggest that, due to the importance of Cabo Verde as a commercial entrepôt in the Atlantic slave trade, establishing the precise place of origin of this introduced species is challenging. Non-invasive fecal samples were collected from feral and captive green monkey individuals in Cabo Verde. Two mitochondrial fragments, HVRI and cyt b, were used to confirm the taxonomic identification of the species and to tentatively determine the geographic origin of introduction to the archipelago from the African continent. By comparing the new sequences of this study to previously published ones, it was shown that Cabo Verde individuals have unique haplotypes in the HVRI, while also showing affinities to several populations from north-western coastal Africa in the cyt b, suggesting probable multiple sources of introduction and an undetermined most probable origin. The latter is consistent with historical information, but may also have resulted from solely using mtDNA as a genetic marker and the dispersal characteristics of the species. The limitations of the methodology are discussed and future directions of research are suggested.
Full article
(This article belongs to the Section Animal Genetics and Genomics)




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}