





Genes 2024, 15(5), 529; https://doi.org/10.3390/genes15050529 - 23 Apr 2024
Abstract
The evolutionary conserved Notch signaling pathway functions as a mediator of direct cell–cell communication between neighboring cells during development. Notch plays a crucial role in various fundamental biological processes in a wide range of tissues. Accordingly, the aberrant signaling of this pathway underlies
[...] Read more.
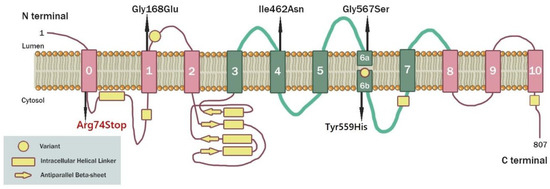
The evolutionary conserved Notch signaling pathway functions as a mediator of direct cell–cell communication between neighboring cells during development. Notch plays a crucial role in various fundamental biological processes in a wide range of tissues. Accordingly, the aberrant signaling of this pathway underlies multiple genetic pathologies such as developmental syndromes, congenital disorders, neurodegenerative diseases, and cancer. Over the last two decades, significant data have shown that the Notch signaling pathway displays a significant function in the mature brains of vertebrates and invertebrates beyond neuronal development and specification during embryonic development. Neuronal connection, synaptic plasticity, learning, and memory appear to be regulated by this pathway. Specific mutations in human Notch family proteins have been linked to several neurodegenerative diseases including Alzheimer’s disease, CADASIL, and ischemic injury. Neurodegenerative diseases are incurable disorders of the central nervous system that cause the progressive degeneration and/or death of brain nerve cells, affecting both mental function and movement (ataxia). There is currently a lot of study being conducted to better understand the molecular mechanisms by which Notch plays an essential role in the mature brain. In this study, an in silico analysis of polymorphisms and mutations in human Notch family members that lead to neurodegenerative diseases was performed in order to investigate the correlations among Notch family proteins and neurodegenerative diseases. Particular emphasis was placed on the study of mutations in the Notch3 protein and the structure analysis of the mutant Notch3 protein that leads to the manifestation of the CADASIL syndrome in order to spot possible conserved mutations and interpret the effect of these mutations in the Notch3 protein structure. Conserved mutations of cysteine residues may be candidate pharmacological targets for the potential therapy of CADASIL syndrome.
Full article
(This article belongs to the Special Issue Head and Neck Genetics)








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
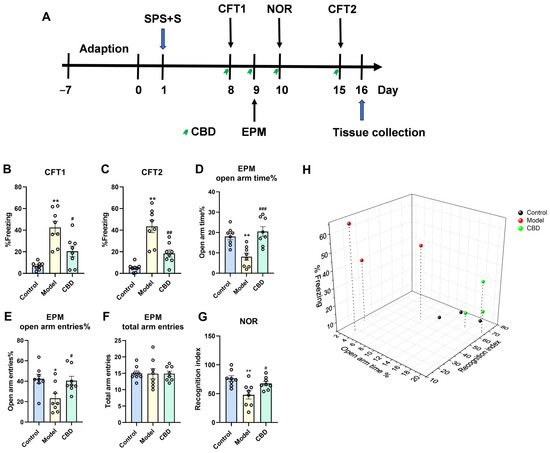{kind=link}
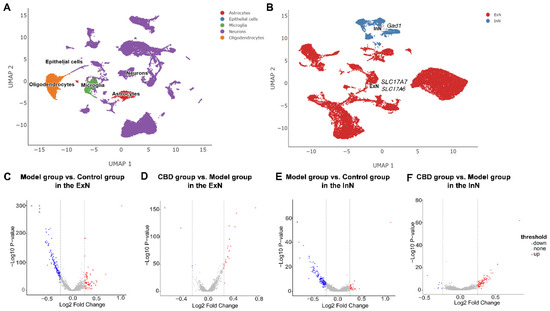{kind=link}
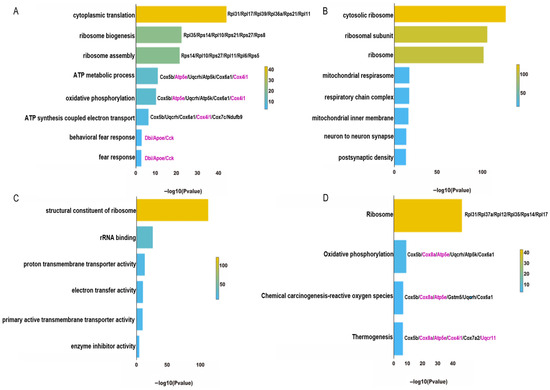{kind=link}
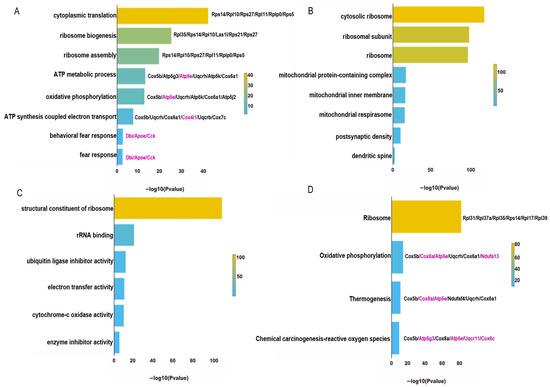{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}