





Genes 2024, 15(4), 511; https://doi.org/10.3390/genes15040511 - 18 Apr 2024
Abstract
The loss of anthocyanin pigments is one of the most common evolutionary transitions in petal color, yet the genetic basis for these changes in flax remains largely unknown. In this study, we used crossing studies, a bulk segregant analysis, genome-wide association studies, a
[...] Read more.
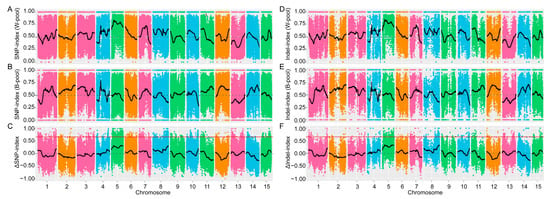
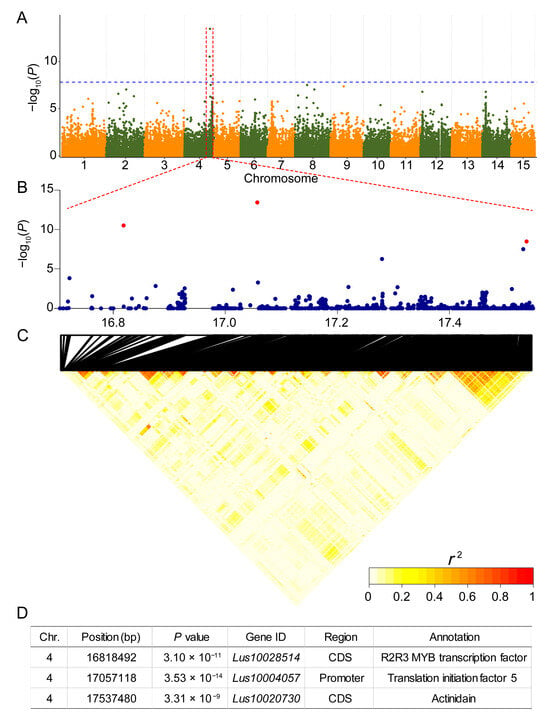
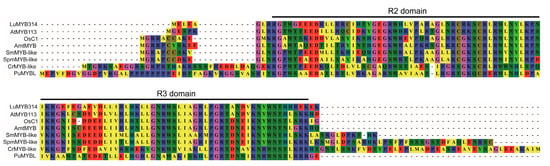
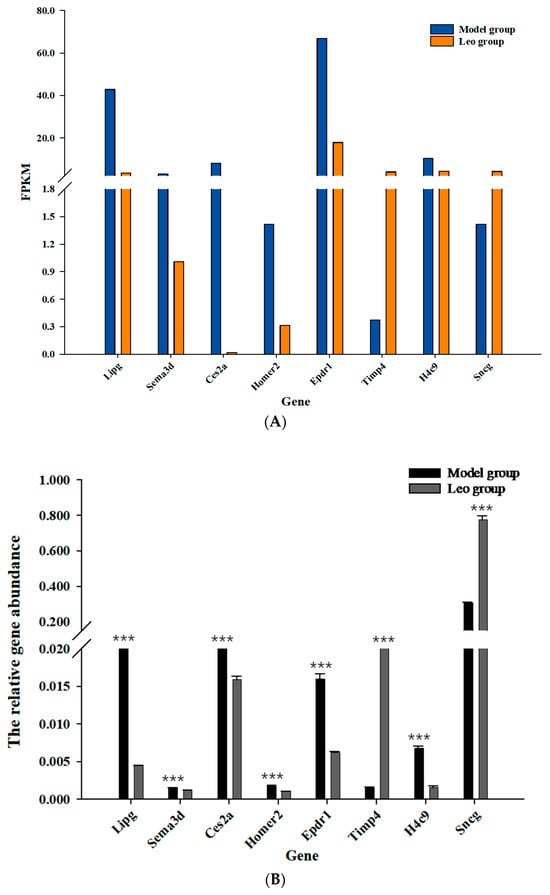
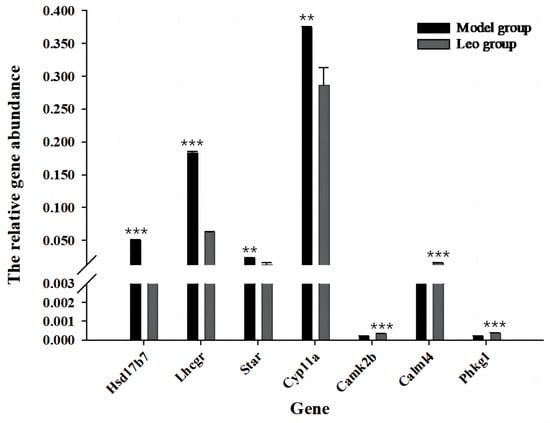
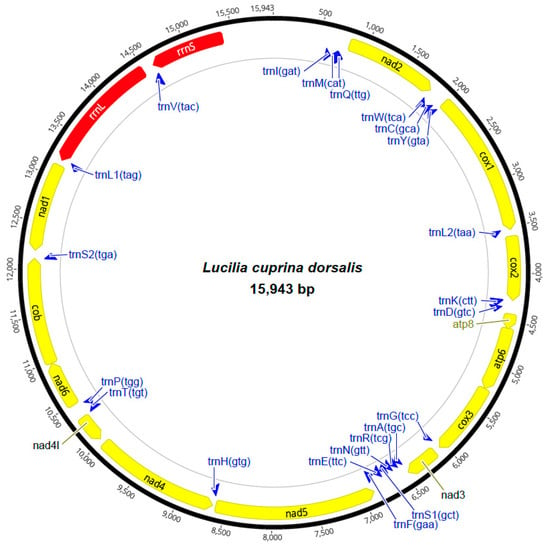
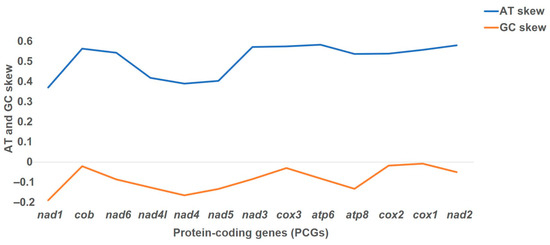
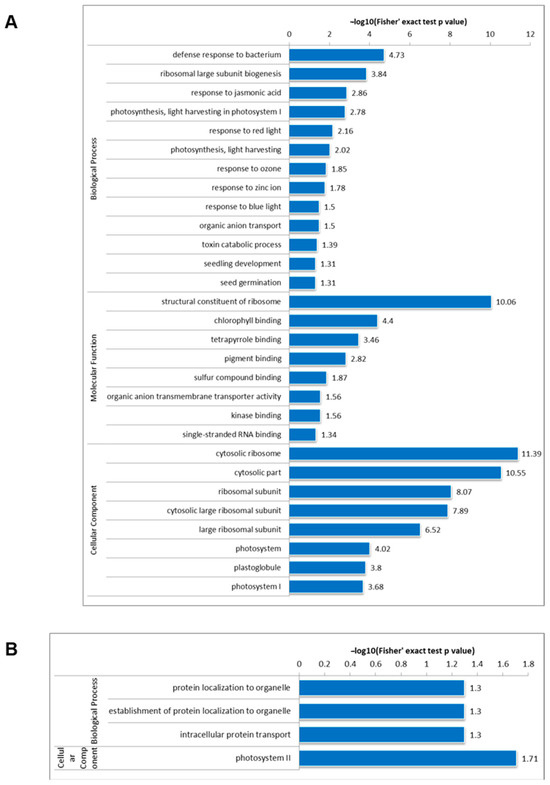
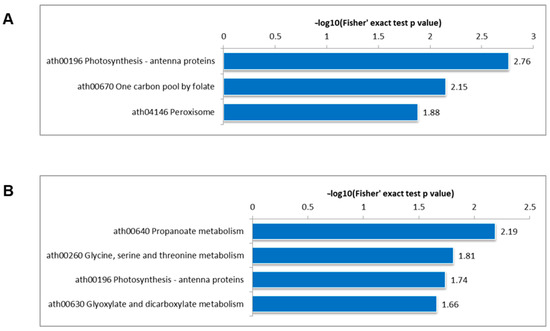
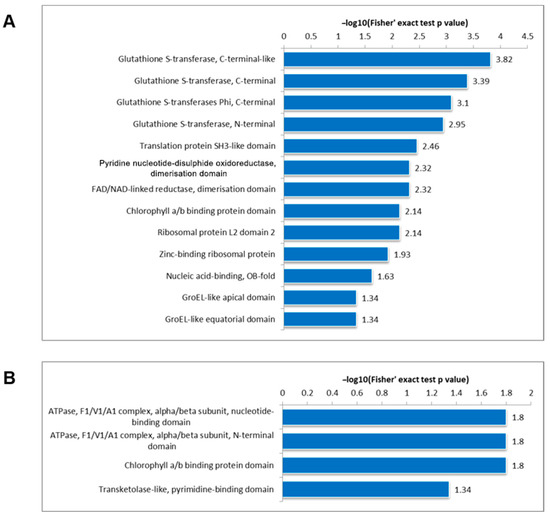
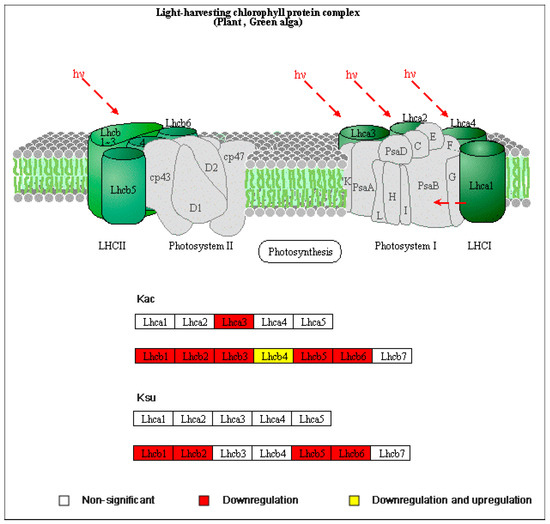
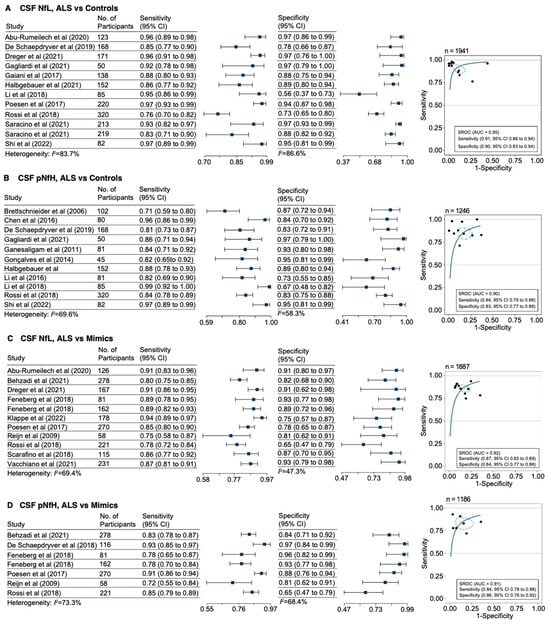
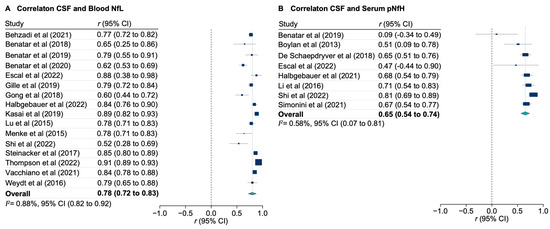
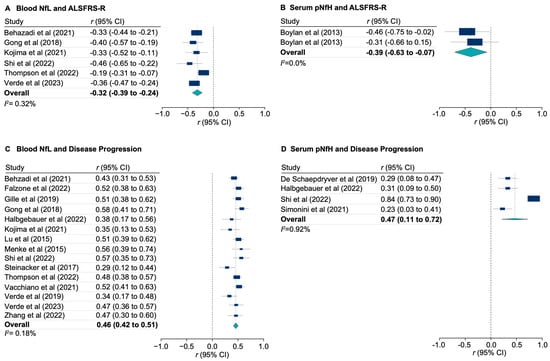
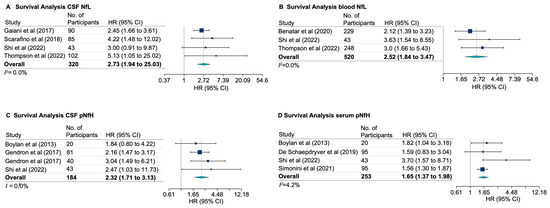
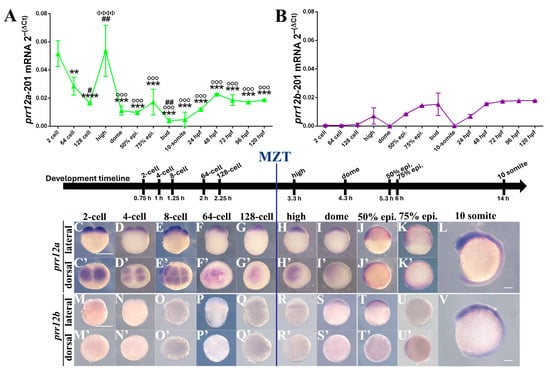
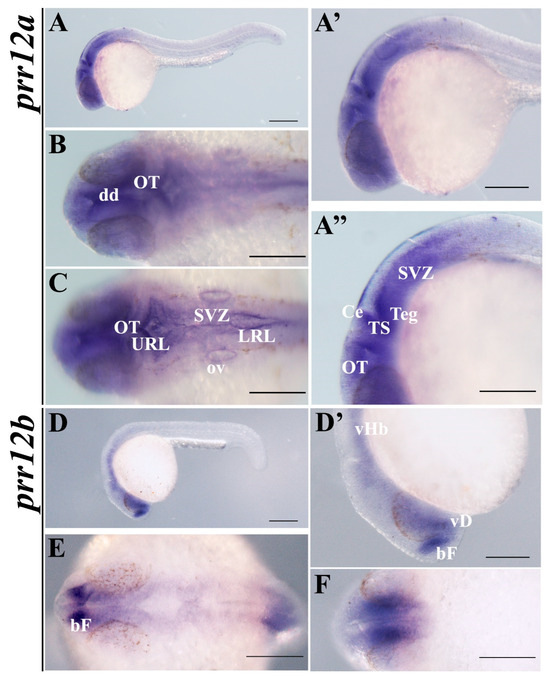
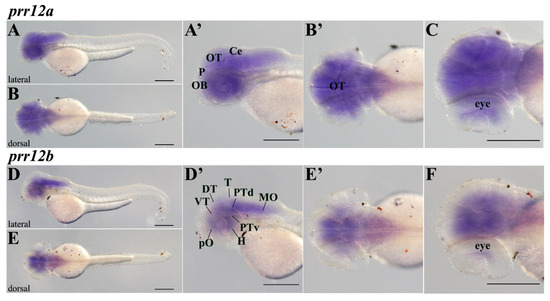
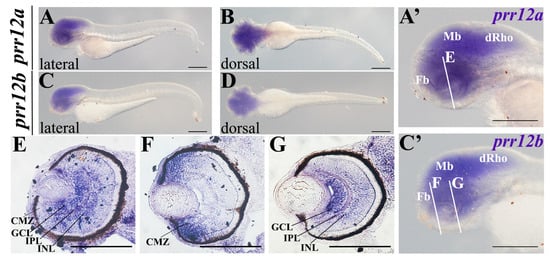
The loss of anthocyanin pigments is one of the most common evolutionary transitions in petal color, yet the genetic basis for these changes in flax remains largely unknown. In this study, we used crossing studies, a bulk segregant analysis, genome-wide association studies, a phylogenetic analysis, and transgenic testing to identify genes responsible for the transition from blue to white petals in flax. This study found no correspondence between the petal color and seed color, refuting the conclusion that a locus controlling the seed coat color is associated with the petal color, as reported in previous studies. The locus controlling the petal color was mapped using a BSA-seq analysis based on the F2 population. However, no significantly associated genomic regions were detected. Our genome-wide association study identified a highly significant QTL (BP4.1) on chromosome 4 associated with flax petal color in the natural population. The combination of a local Manhattan plot and an LD heat map identified LuMYB314, an R2R3-MYB transcription factor, as a potential gene responsible for the natural variations in petal color in flax. The overexpression of LuMYB314 in both Arabidopsis thaliana and Nicotiana tabacum resulted in anthocyanin deposition, indicating that LuMYB314 is a credible candidate gene for controlling the petal color in flax. Additionally, our study highlights the limitations of the BSA-seq method in low-linkage genomic regions, while also demonstrating the powerful detection capabilities of GWAS based on high-density genomic variation mapping. This study enhances our genetic insight into petal color variations and has potential breeding value for engineering LuMYB314 to develop colored petals, bast fibers, and seeds for multifunctional use in flax.
Full article
(This article belongs to the Special Issue Advances in Genetics and Genomics of Plants)
►
Show Figures

Figure 1





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}